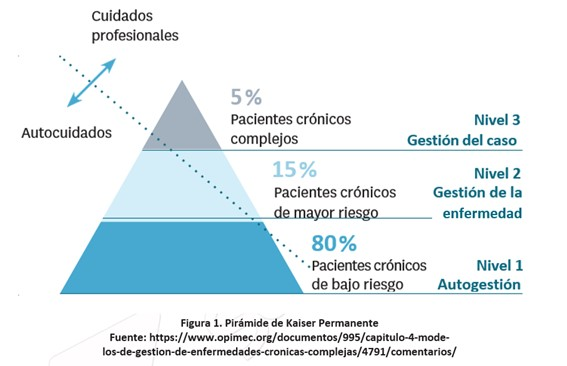
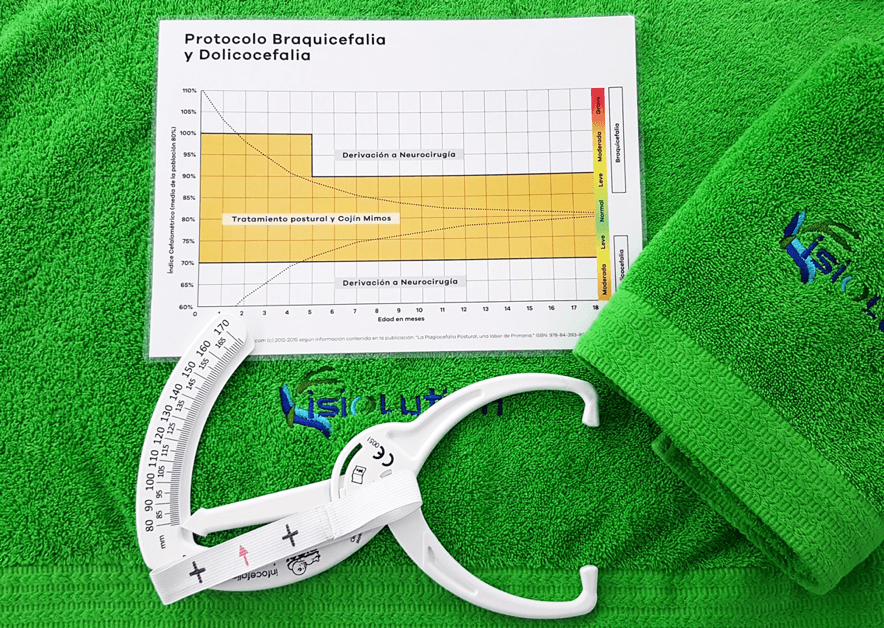
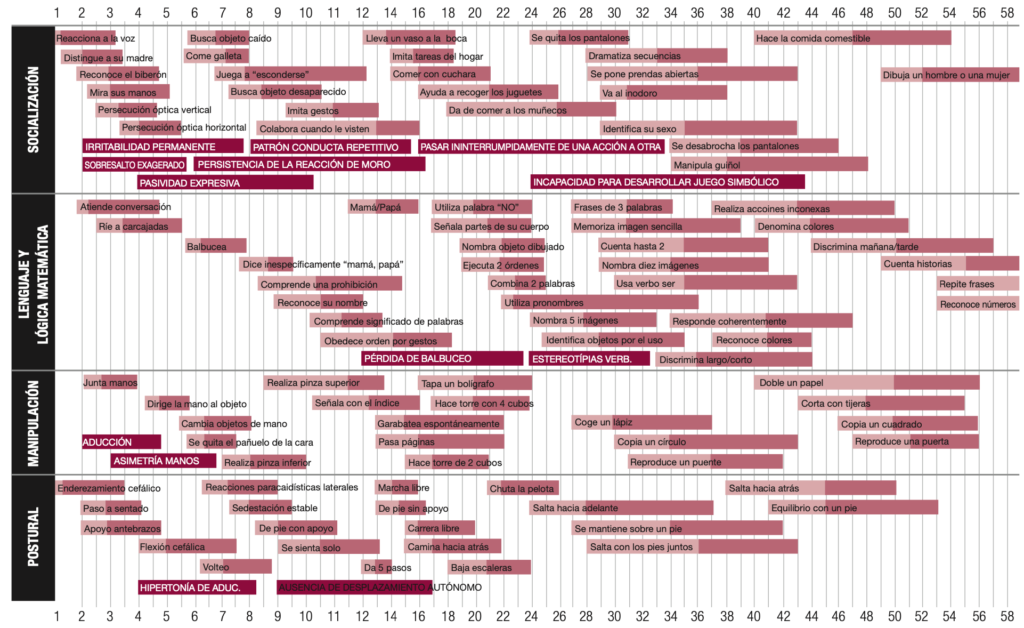
Escalas y calculadoras Application nutritional NIHSS-Ped PedCom Indice cefalométrico y braquicefálico Método de Weaver Test de Haizea-Llevant Calculadora de puntuaciones normalizadas Aplicaciones móviles Normscales Smart Optometry Genética Phenomyzer Decipher Rarecrhomo Varsome Gene Reviews Enfermedades neurológicas Learning EEG Neuromuscular diseases, Washington Epilepsy Diagnosis Movement Disorder Genes Neurochecklists Metabólicas Vademecum Metabolicum Treatable ID Neurorradiologia MRI of the Neonatal Brain Radiopaedia Manejo terapéutico. OrphanAnesthesia Treatabolome