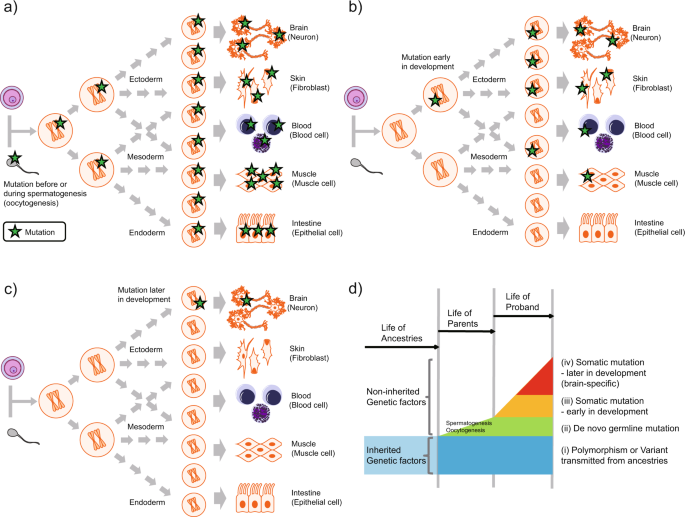
In recent years, "de novo" mutations, those not inherited from either parent, have gained special relevance in pediatric neurology, for several reasons:
They are responsible for the majority of cases of genetically identifiable neurodevelopmental disorders.
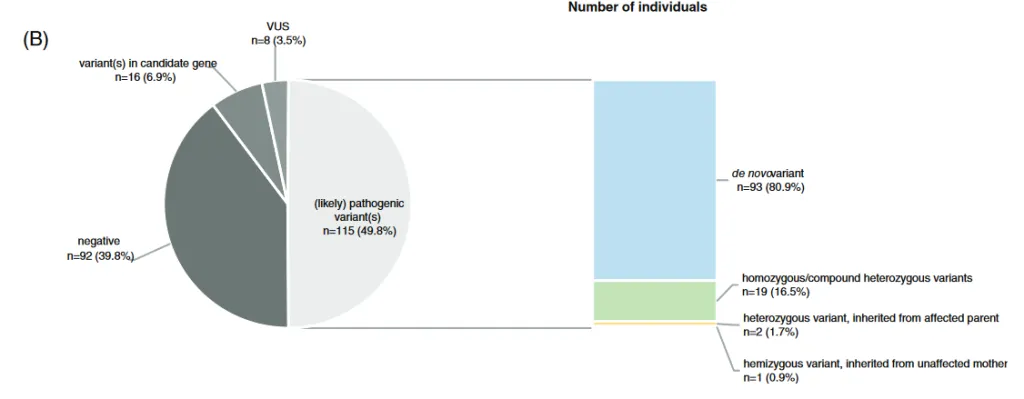
They are responsible for the most serious cases.
This is explained because there is bias due to natural selection. Inherited diseases are usually less serious than "de novo" diseases, because they imply that the individual has been able to live to adulthood, mate, and reproduce. In this way, variants that remain in the general population, and are frequently found in healthy carriers, produce less phenotypic impact, although the fact that a variant is "de novo" does not guarantee pathogenicity.
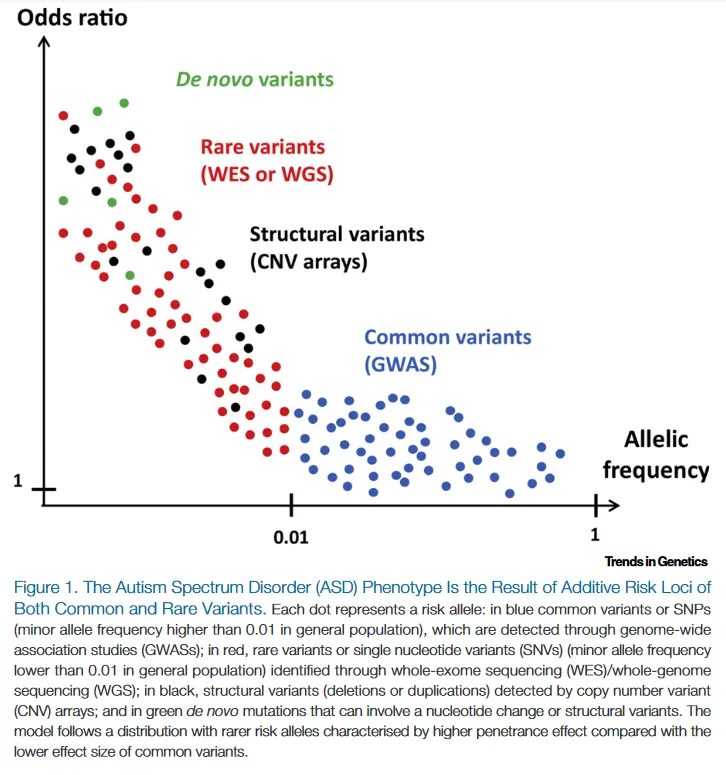
They question the classical paradigm of genetic disease as an inherited disease.
This calls into question the relative importance of family history when it comes to suspecting a genetic disease, since its absence is not a criterion for exclusion.
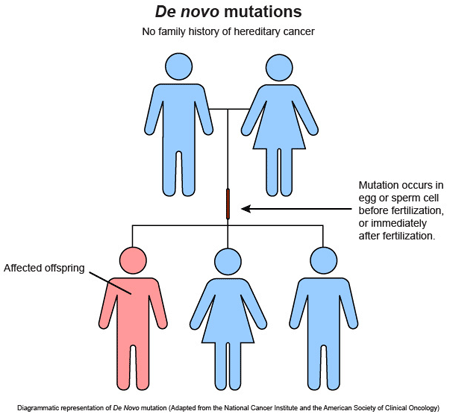
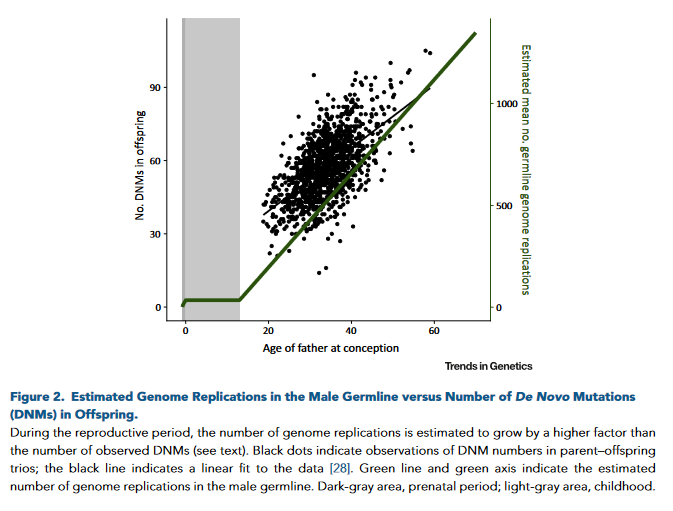
The risk of de novo mutation depends on the parental age.
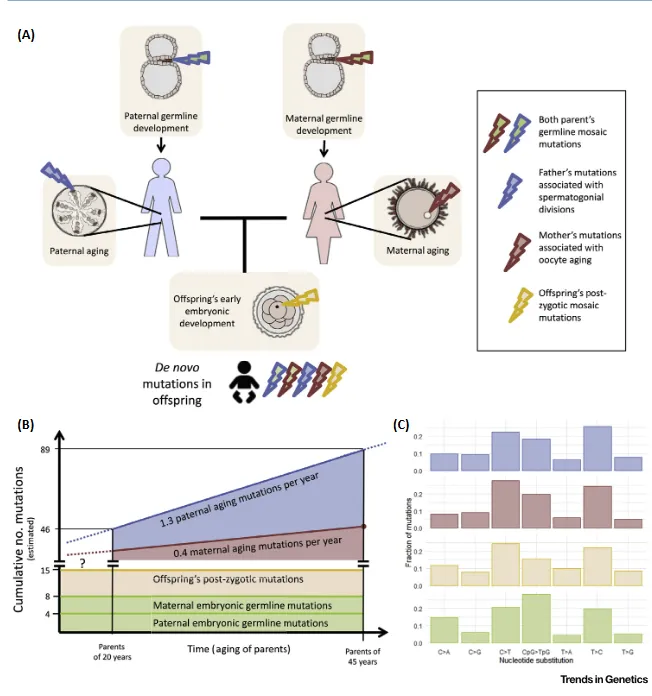
Through genomic studies, we are currently aware that healthy people carry around 50-100 de novo mutations.
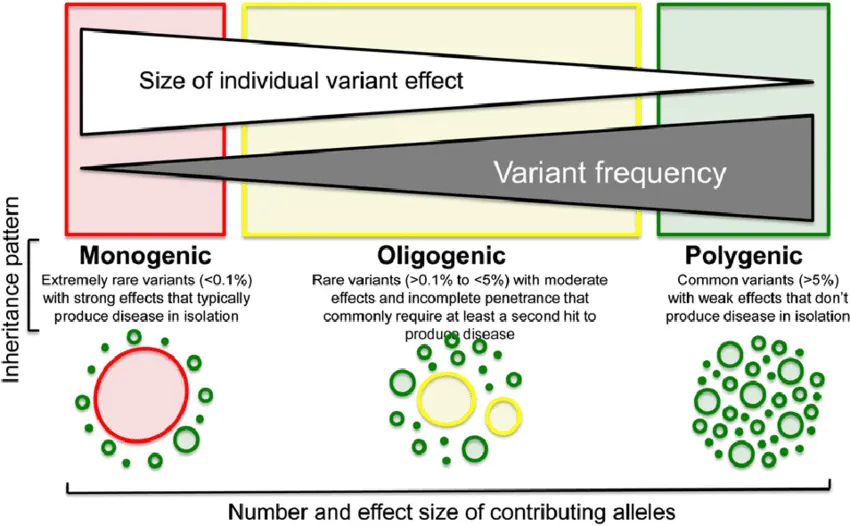
Therefore, it is not uncommon for there to be cases of complex phenotype in which multiple mutations give rise to overlapping phenotypes of greater severity (Double Trouble), giving rise to the concept of "mutational load."
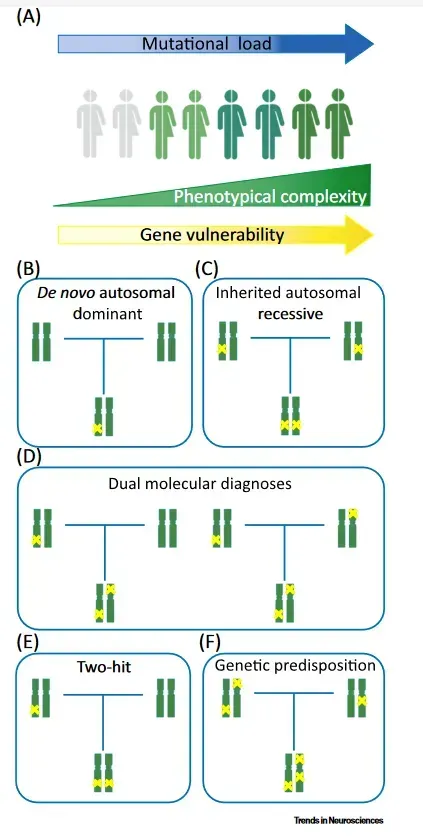
This questions the classic paradigm of monogenetic disease.
In this way, the role of the genetic variants that each individual presents may represent a polygenic inheritance (quantitative, risk factor) when the variants produce little phenotypic impact, or a monogenetic disease, when the location and consequences of the variant produce a serious clinical impact.
They can occur beyond conception, so they are the cause of constitutional mosaicism.