
Bajo el concepto de secuenciación masiva o NGS (next generation sequencing) existen múltiples técnicas diagnósticas de elevada complejidad que han incrementado significativamente las posibilidades de diagnóstico en neurología pediátrica. Podemos clasificarlas en función de varios criterios.
Criterio histórico (generación).

332710
{332710:WU8PQYII}
1
vancouver
50
default
2240
https://neuropediatoolkit.org/wp-content/plugins/zotpress/
%7B%22status%22%3A%22success%22%2C%22updateneeded%22%3Afalse%2C%22instance%22%3Afalse%2C%22meta%22%3A%7B%22request_last%22%3A0%2C%22request_next%22%3A0%2C%22used_cache%22%3Atrue%7D%2C%22data%22%3A%5B%7B%22key%22%3A%22WU8PQYII%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Gupta%20and%20Verma%22%2C%22parsedDate%22%3A%222019%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BGupta%20N%2C%20Verma%20VK.%20Next-Generation%20Sequencing%20and%20Its%20Application%3A%20Empowering%20in%20Public%20Health%20Beyond%20Reality.%20In%3A%20Arora%20PK%2C%20editor.%20Microbial%20Technology%20for%20the%20Welfare%20of%20Society%20%5BInternet%5D.%20Singapore%3A%20Springer%3B%202019%20%5Bcited%202021%20May%202%5D.%20p.%20313%26%23x2013%3B41.%20%28Microorganisms%20for%20Sustainability%29.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2F978-981-13-8844-6_15%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2F978-981-13-8844-6_15%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22bookSection%22%2C%22title%22%3A%22Next-Generation%20Sequencing%20and%20Its%20Application%3A%20Empowering%20in%20Public%20Health%20Beyond%20Reality%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nidhi%22%2C%22lastName%22%3A%22Gupta%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Vijay%20K.%22%2C%22lastName%22%3A%22Verma%22%7D%2C%7B%22creatorType%22%3A%22editor%22%2C%22firstName%22%3A%22Pankaj%20Kumar%22%2C%22lastName%22%3A%22Arora%22%7D%5D%2C%22abstractNote%22%3A%22Next-generation%20sequencing%20has%20the%20ability%20to%20revolutionize%20almost%20all%20fields%20of%20biological%20science.%20It%20has%20drastically%20reduced%20the%20cost%20of%20sequencing.%20This%20allows%20us%20to%20study%20the%20whole%20genome%20or%20part%20of%20the%20genome%20to%20understand%20how%20the%20cellular%20functions%20are%20governed%20by%20the%20genetic%20code.%20The%20data%20obtained%20in%20huge%20quantity%20from%20sequencing%20upon%20analysis%20gives%20an%20insight%20to%20understand%20the%20mechanism%20of%20pathogen%20biology%2C%20virulence%2C%20and%20phenomenon%20of%20bacterial%20resistance%2C%20which%20helps%20in%20investigating%20the%20outbreak.%20This%20ultimately%20helps%20in%20the%20development%20of%20therapies%20for%20public%20health%20welfare%20against%20human%20pathogen%20and%20diagnostic%20reagents%20for%20the%20screening.%20This%20chapter%20includes%20the%20basic%20of%20Sanger%5Cu2019s%20method%20of%20DNA%20sequencing%20and%20next-generation%20sequencing%2C%20different%20available%20platforms%20for%20sequencing%20with%20their%20advantages%2C%20and%20limitations%20and%20their%20chemistry%20with%20an%20overview%20of%20downstream%20data%20analysis.%20Furthermore%2C%20the%20breadth%20of%20applications%20of%20high-throughput%20NGS%20technology%20for%20human%20health%20has%20been%20discussed.%22%2C%22bookTitle%22%3A%22Microbial%20Technology%20for%20the%20Welfare%20of%20Society%22%2C%22date%22%3A%222019%22%2C%22originalDate%22%3A%22%22%2C%22originalPublisher%22%3A%22%22%2C%22originalPlace%22%3A%22%22%2C%22format%22%3A%22%22%2C%22ISBN%22%3A%22978-981-13-8844-6%22%2C%22DOI%22%3A%2210.1007%5C%2F978-981-13-8844-6_15%22%2C%22citationKey%22%3A%22%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2F978-981-13-8844-6_15%22%2C%22ISSN%22%3A%22%22%2C%22language%22%3A%22en%22%2C%22collections%22%3A%5B%227XAESF88%22%5D%2C%22dateModified%22%3A%222025-09-27T20%3A43%3A24Z%22%7D%7D%5D%7D
1.
Gupta N, Verma VK. Next-Generation Sequencing and Its Application: Empowering in Public Health Beyond Reality. In: Arora PK, editor. Microbial Technology for the Welfare of Society [Internet]. Singapore: Springer; 2019 [cited 2021 May 2]. p. 313–41. (Microorganisms for Sustainability). Available from: https://doi.org/10.1007/978-981-13-8844-6_15
- Los métodos de secuenciación de segunda generación se basan en el principio del «shotgun sequencing«, y requieren la fragmentación del DNA en estudio, pero lamentablemente la longitud de las lecturas es corta (short-read sequencing), por lo que determinados tipos de mutaciones no son identificables (STR, delecciones o duplicaciones, etc.).
- Los métodos de tercera y cuarta generación se han desarrollado con el objetivo de superar esta limitación, por lo que también se llaman long-read sequencing.
Tipo de captura.
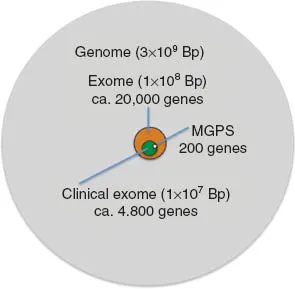
El tipo de captura utilizado es de una importancia clínica fundamental, ya que condicionará casi más que el tipo de tecnología utilizado, las decisiones clínicas que deberemos tomar en caso de no obtener resultados diagnósticos definitivos.
Panel de genes.
- Implica el análisis de un máximo de 200 genes aproximadamente.
- Permite el estudio de enfermedades con variabilidad genotípica, pero requiere de la generación de una hipótesis previa (phenotype first approach).
- Limita significativamente el número de hallazgos no relacionados con el problema a estudio, así como las variantes de significado incierto.
Estudio de secuenciación del exoma clínico/dirigido/mendelioma
- Consiste en el análisis de todas las regiones codificantes (las regiones codificantes suponen el 1% del genoma, y acumulan el 80% de las variantes patogénicas conocidas) para las que se han descrito previamente genes relacionados con enfermedad (aquellos genes con función desconocida no aportan información clínicamente relevante aún cuando descubramos variantes patogénicas).
- Permite el estudio sin plantear una hipótesis previa (genotype first approach), y implica el análisis de más de 4000 genes (dependiendo de la evolución del conocimiento científico, actualmente en OMIM ya superan los 6000), lo que equivale a un tamaño de unas 10 Mb.
- Permite la detección de variantes no codificantes flanqueantes (variantes en 3′ o 5′ UTR).
- No permite el uso con fines de investigación de genes con función no conocida, ya que no se incluyen, ni permitirá por tanto el reanálisis de dichos genes, ya que no se ha obtenido su secuencia.
- El reanálisis de los datos tras varios años puede ser de interés, ya que el número de genes estudiado hace que puedan no haberse descrito variantes patogénicas que serán descubiertas en un futuro.
- Ofrece un buen equilibrio entre el riesgo de variantes de significado incierto y la rentabilidad diagnóstica sin disparar los costes económicos y de trabajo del análisis de un exoma completo o un genoma.
Estudio de secuenciación del exoma.
- Permite la detección de variantes en las regiones codificantes para proteínas, tanto en genes ya relacionados con patología como en aquellos en los que no se ha descrito, por lo que implica el análisis de más de 20.000 genes, lo que equivale a un tamaño de unas 100 Mb.
- Permite la detección de variantes no codificantes flanqueantes (variantes en 3′ o 5′ UTR).
- Permite la formulación de hipótesis de investigación, y el descubrimiento de nuevas enfermedades y de funciones no conocidas de genes que actualmente aún no están bien descritos.
- Permite el reanálisis de los datos en busca de nuevo conocimiento tras varios años desde la realización del estudio sin necesidad de obtener nuevas muestras.
- Eleva significativamente el coste en términos económicos y en trabajo.
- Eleva significativamente la probabilidad de hallazgos de significado incierto y hallazgos inesperados, por lo que es recomendable la realización del estudio en trio para cruzar la información familiar de forma automatizada.
Estudio de secuenciación del genoma.
- Permite la detección de todas las variantes en regiones codificantes para proteínas.
- Permite la detección todas las variantes no codificantes (intrónicas profundas, variantes en 3′ o 5′ UTR).
- Permite la detección de variantes en regiones reguladoras (enhacers, silencers, insulators).
- Permite la detección de pequeñas delecciones o duplicaciones (de exón único).
- Permite la detección de variantes estructurales (translocaciones, inserciones, delecciones, y sus puntos de ruptura).
- Es el proceso más costoso en términos económico y en trabajo.
- Eleva significativamente la probabilidad de hallazgos de significado incierto y hallazgos inesperados, por lo que es recomendable la realización del estudio en trio para cruzar la información familiar de forma automatizada.
- Equivale aproximadamente a un tamaño de 3 Gb.
Puedes consultar los distintos tipos de captura comercializados para conocer su extensión.
Tipo de tecnología de secuenciación

332710
{332710:WU8PQYII}
1
vancouver
50
default
2240
https://neuropediatoolkit.org/wp-content/plugins/zotpress/
%7B%22status%22%3A%22success%22%2C%22updateneeded%22%3Afalse%2C%22instance%22%3Afalse%2C%22meta%22%3A%7B%22request_last%22%3A0%2C%22request_next%22%3A0%2C%22used_cache%22%3Atrue%7D%2C%22data%22%3A%5B%7B%22key%22%3A%22WU8PQYII%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Gupta%20and%20Verma%22%2C%22parsedDate%22%3A%222019%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BGupta%20N%2C%20Verma%20VK.%20Next-Generation%20Sequencing%20and%20Its%20Application%3A%20Empowering%20in%20Public%20Health%20Beyond%20Reality.%20In%3A%20Arora%20PK%2C%20editor.%20Microbial%20Technology%20for%20the%20Welfare%20of%20Society%20%5BInternet%5D.%20Singapore%3A%20Springer%3B%202019%20%5Bcited%202021%20May%202%5D.%20p.%20313%26%23x2013%3B41.%20%28Microorganisms%20for%20Sustainability%29.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2F978-981-13-8844-6_15%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2F978-981-13-8844-6_15%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22bookSection%22%2C%22title%22%3A%22Next-Generation%20Sequencing%20and%20Its%20Application%3A%20Empowering%20in%20Public%20Health%20Beyond%20Reality%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nidhi%22%2C%22lastName%22%3A%22Gupta%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Vijay%20K.%22%2C%22lastName%22%3A%22Verma%22%7D%2C%7B%22creatorType%22%3A%22editor%22%2C%22firstName%22%3A%22Pankaj%20Kumar%22%2C%22lastName%22%3A%22Arora%22%7D%5D%2C%22abstractNote%22%3A%22Next-generation%20sequencing%20has%20the%20ability%20to%20revolutionize%20almost%20all%20fields%20of%20biological%20science.%20It%20has%20drastically%20reduced%20the%20cost%20of%20sequencing.%20This%20allows%20us%20to%20study%20the%20whole%20genome%20or%20part%20of%20the%20genome%20to%20understand%20how%20the%20cellular%20functions%20are%20governed%20by%20the%20genetic%20code.%20The%20data%20obtained%20in%20huge%20quantity%20from%20sequencing%20upon%20analysis%20gives%20an%20insight%20to%20understand%20the%20mechanism%20of%20pathogen%20biology%2C%20virulence%2C%20and%20phenomenon%20of%20bacterial%20resistance%2C%20which%20helps%20in%20investigating%20the%20outbreak.%20This%20ultimately%20helps%20in%20the%20development%20of%20therapies%20for%20public%20health%20welfare%20against%20human%20pathogen%20and%20diagnostic%20reagents%20for%20the%20screening.%20This%20chapter%20includes%20the%20basic%20of%20Sanger%5Cu2019s%20method%20of%20DNA%20sequencing%20and%20next-generation%20sequencing%2C%20different%20available%20platforms%20for%20sequencing%20with%20their%20advantages%2C%20and%20limitations%20and%20their%20chemistry%20with%20an%20overview%20of%20downstream%20data%20analysis.%20Furthermore%2C%20the%20breadth%20of%20applications%20of%20high-throughput%20NGS%20technology%20for%20human%20health%20has%20been%20discussed.%22%2C%22bookTitle%22%3A%22Microbial%20Technology%20for%20the%20Welfare%20of%20Society%22%2C%22date%22%3A%222019%22%2C%22originalDate%22%3A%22%22%2C%22originalPublisher%22%3A%22%22%2C%22originalPlace%22%3A%22%22%2C%22format%22%3A%22%22%2C%22ISBN%22%3A%22978-981-13-8844-6%22%2C%22DOI%22%3A%2210.1007%5C%2F978-981-13-8844-6_15%22%2C%22citationKey%22%3A%22%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2F978-981-13-8844-6_15%22%2C%22ISSN%22%3A%22%22%2C%22language%22%3A%22en%22%2C%22collections%22%3A%5B%227XAESF88%22%5D%2C%22dateModified%22%3A%222025-09-27T20%3A43%3A24Z%22%7D%7D%5D%7D
1.
Gupta N, Verma VK. Next-Generation Sequencing and Its Application: Empowering in Public Health Beyond Reality. In: Arora PK, editor. Microbial Technology for the Welfare of Society [Internet]. Singapore: Springer; 2019 [cited 2021 May 2]. p. 313–41. (Microorganisms for Sustainability). Available from: https://doi.org/10.1007/978-981-13-8844-6_15
- Es de interés conocer que los métodos que utilizan amplificación, pueden inducir un sesgo en el análisis de las secuencias ricas en GC, por las dificultades que encuentra la PCR en estas regiones.
https://link.springer.com/chapter/10.1007/978-981-13-8844-6_15