
Los informes de resultados de secuenciación son documentos complejos llenos de datos alfanuméricos dificiles de entender sin unas bases generales que nos ayuden a interpretarlos. Habitualmente nos centramos en la conclusión final, es decir, el hallazgo de una variante genética determinada y su interpretación de patogenicidad, es la parte de información más relevante clínicamente, pero podemos obtener mucha más información si somos capaces de entender el cuerpo de texto y hacer uso de esta en las distintas bases de datos informatizadas.
Para ello hemos de ser conocedores de que las variantes genéticas identificadas por secuenciación hacen referencia a una posición de una base nitrogenada en el genoma (unas coordenadas). Para poder posicionar dicha base es necesario conocer el marco de referencia en el que nos basamos (desde donde contamos la posición). Este marco de referencia depende de varios factores.
Nomenclatura. HGVS.
En primer lugar deberemos entender que cambio se ha producido en el DNA, y que consecuencias produce en la proteína. Existe una nomenclatura internacional para hacer referencia a dicho cambio. El organismo internacional encargado de fijar las abreviaturas y términos estandarizados para referirse a una variante o mutación puntual se llama Human Genome Variation Society, y puede consultarse en internet http://varnomen.hgvs.org/
Existen incluso herramientas informáticas que permiten la codificación en nomenclatura estándar, a partir de la secuencia de nucleótidos y la secuencia estándar con la que se compara.
Las principales reglas utilizadas para nombrar una variante genética son las siguientes:
En primer lugar, para evitar confusión, la variante reportada debe de ser precedida por una letra que indique el tipo de DNA al que se refiere:
- «c.» para una secuencia de ADN codificante. (como c.76A>T)
- «g.» para una secuencia genómica (como g.476A>T)
- «m.» para una secuencia mitocondrial (como m.8993T>C)
- «n.» para un ARN no codificante (un gen que produce un transcrito ARN pero no una proteína).
- «r.» para una secuencia ARN (como r.76a>u)
- «p.» para una secuencia de proteína (como p.Lys76Asn)
En segundo lugar, sigue un número que indica la posición del nucléotido o el aminoácido que se ha alterado en la secuencia de referencia.
- c.76A>T. en la posición 76 una adenina se ha modificado por una timina.
- p.Arg1543>Lys. En la posición 1543 una arginina se ha modificado por una lisina.
En tercer lugar, es necesario indicar los cambios específicos identificados en la secuencia.
- «>» indica una sustitución a nivel del ADN (como c.76A>T)
- «_» (guión bajo) indica un rango de resíduos afectado, separando el primer y el último resíduo de dicho rango (como c.76_78delACT)
- «del» indica una delección (como c.76delA)
- «dup» indica una duplicación (como c.76dupA);
- «ins» indica una inserción (como c.76_77insG).
- las inserciones que en realidad son una duplicación, se describen como duplicaciones, no como inserciones; el cambio de ACTTTGTGCC a ACTTTGTGGCC se describe como c.8dupG (no como c.8_9insG).
- «inv» indica una inversión (como c.76_83inv)
- «con» indica una conversión (como c.123_678conNM_004006.1:c.123_678)
- «[]» indica un alelo (como c.[76A>T)
- «()» se usa cuando la posición exacta de un cambio no se conoce; el rango de incertidumbre debe ser descrito de forma tan precisa como sea posible y incluido entre los paréntesis (como c.(67_70)insG)
- la variabilidad en el número de secuencias repetidas (como por ejemplo en ATGCGATGTGTGCC) se describen indicando el número de repeticiones y los nucleótidos implicados, así como la posición, como c.123+74TG(3_6)
- las triplicaciones y cuadruplicaciones se describen como alelos de repeticiones de secuencias cortas; c.87_93[3] describe una triplicación de 7 nucleótidos desde la posición codificante 87 a la 93 (no se nombra como c.87_93tri)
Cuando existen múltiples cambios de secuencia en el mismo individuo, deben nombrarse:
- dos cambios en la secuencia localizados en distintos alelos, como los existentes en las enfermedades recesivas, se deben describir entre dos conjuntos de corchetes, separados por «;», como en c.[76A>C];[87delG].
- dos cambios en la secuencia localizados en el mismo alelo, se deben describir dentro del mismo corchete, separados por «;», como en c.[76A>C; 83G>C]
- dos cambios en la secuencia en los que se desconoce su posición en los alelos se deben describir entre corchetes, separados por un punto y coma entre paréntesis “(;)»; c.[76A>C(;)83G>C]
- los cambios de secuencia en diferentes genes deben describirse entre corchetes, separados por «;» e incluir una referencia al gen en el que se encuentra el cambio; DMD:c.[76A>C];GJB:c.[87delG].
- mosaicos: dos nucleótidos diferentes en la misma posición de un mismo alelo se describen entre corchetes, separados por «/»; c.[=/83G>C].
- quimeras: dos nucleótidos diferentes en la misma posición de un mismo alelo se describen entre corchetes, separados por «//»; c.[=//83G>C].
Transcrito.
En segundo lugar tendremos que buscar el tránscrito en el que se encuadra la variante, ya que la posición que identifica al cambio de nucleótido se cuenta desde el inicio de dicho tránscrito. Esto es necesario porque la mayoría de genes dan lugar a varias moléculas de ARN mensajero distintas con longitudes distintas (tránscritos), como mecanismo evolutivo para incrementar la diversidad genética, que a su vez se traduciran finalmente distintas isoformas de la proteína que codifica el gen.
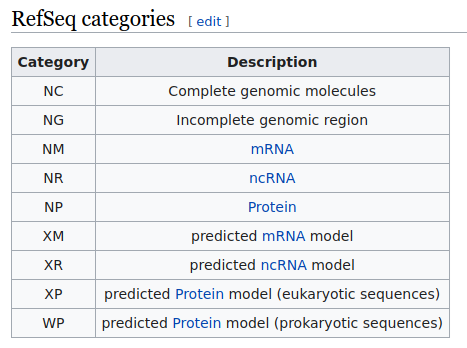
Dado el gran número de tránscritos existentes, ha sido necesario generar bases de datos específicas para poder identificarlos. La más utilizada es RefSeq, que ha indexado todas las moléculas biológicas humanas de origen natural basadas en nucleótidos (ADN, ARN) y asociadas a una proteína, codificadas con un número único para cada registro.
Habitualmente encontraremos el código del tránscrito en el que se basa la posición del cambio de bases en un cuadro de análisis del informe de la variante detectada, precedido por las letras NM (que se refieren a RNA mensajero). Existen otras categorías en RefSeq, como NG o NC.
Genoma de referencia.
En tercer lugar, deberemos conocer el genoma de referencia que ha sido utilizado para el análisis, puesto que el avance del conocimiento científico conlleva que vayan realizándose actualizaciones de dicho genoma de referencia. El consorcio del genoma de referencia es la entidad encargada de mantener, revisar y aceptar las modificaciones en el genoma de referencia que se van proponiendo por parte de la comunidad científica internacional, realizándose actualizaciones en versiones sucesivas de dicho genoma de referencia. Actualmente se está utilizando la GRCh38 (Genome Reference Consortium human 38).
En las versiones sucesivas se han ido incorporando variantes existentes en las distintas poblaciones humanas, con el objetivo de representar con la mayor exactitud la diversidad genética global, por lo que el proyecto ha ido incrementándose progresivamente en complejidad.
Encontraremos esta información habitualmente en los apéndices del informe, incluida dentro del capítulo dedicado a la metodología.