
Sequencing results reports are complex documents full of alphanumeric data that are difficult to understand without general bases to help us interpret them. We usually focus on the final conclusion, that is, the finding of a specific genetic variant and its interpretation of pathogenicity, which is the most clinically relevant part of the information, but we can obtain much more information if we are able to understand the body of text and make use of it in the different computerized databases.
To do this we must be aware that the genetic variants identified by sequencing refer to a position of a nitrogenous base in the genome (coordinates). In order to position said base, it is necessary to know the frame of reference on which we base ourselves (from where we count the position). This frame of reference depends on several factors.
Nomenclature. HGVS.
First of all, we must understand what change has occurred in the DNA, and what consequences it produces in the protein. There is an international nomenclature to refer to this change. The international organization in charge of establishing standardized abbreviations and terms to refer to a variant or point mutation is called the Human Genome Variation Society, and can be consulted on the internet http://varnomen.hgvs.org/
There are even computer tools that allow coding in standard nomenclature, based on the nucleotide sequence and the standard sequence with which it is compared.
The main rules used to name a genetic variant are the following:
First of all, to avoid confusion, the reported variant must be preceded by a letter that indicates the type of DNA to which it refers:
- "c." for a coding DNA sequence. (as c.76A>T)
- "g." for a genomic sequence (such as g.476A>T)
- "m." for a mitochondrial sequence (such as m.8993T>C)
- "n." for a non-coding RNA (a gene that produces an RNA transcript but not a protein).
- "r." for an RNA sequence (such as r.76a>u)
- "p." for a protein sequence (such as p.Lys76Asn)
Second, it is followed by a number indicating the position of the nucleotide or amino acid that has been altered in the reference sequence.
- c.76A>T. at position 76 an adenine has been modified by a thymine.
- p.Arg1543>Lys. At position 1543 an arginine has been modified by a lysine.
Third, it is necessary to indicate the specific changes identified in the sequence.
- ">" indicates a substitution at the DNA level (such as c.76A>T)
- «_» (underscore) indicates an affected residue range, separating the first and last residue of said range (such as c.76_78delACT)
- “del” indicates a deletion (such as c.76delA)
- “dup” indicates a duplication (such as c.76dupA);
- "ins" indicates an insertion (such as c.76_77insG).
- insertions that are actually a duplication are described as duplications, not insertions; the change from ACTTTGTGCC to ACTTTGTGGCC is described as c.8dupG (not c.8_9insG).
- "inv" indicates an inversion (such as c.76_83inv)
- "with" indicates a conversion (such as c.123_678conNM_004006.1:c.123_678)
- «[]» indicates an allele (such as c.[76A>T)
- "()" is used when the exact position of a change is not known; the uncertainty range should be described as precisely as possible and included in parentheses (such as c.(67_70)insG)
- the variability in the number of repeated sequences (as for example in ATGCGATGTGTGCC) are described indicating the number of repeats and the nucleotides involved, as well as the position, such as c.123+74TG(3_6)
- triplications and quadruplications are described as alleles of short sequence repeats; c.87_93[3] describes a triplication of 7 nucleotides from coding position 87 to 93 (not named c.87_93tri)
When there are multiple sequence changes in the same individual, they should be named:
- Two sequence changes located on different alleles, such as those found in recessive diseases, must be described between two sets of square brackets, separated by ";", as in c.[76A>C];[87delG].
- two sequence changes located in the same allele must be described within the same square bracket, separated by ";", as in c.[76A>C; 83G>C]
- two sequence changes where their position on the alleles is unknown should be described in square brackets, separated by a semicolon in parentheses “(;)”; c.[76A>C(;)83G>C]
- Sequence changes in different genes should be described in square brackets, separated by ";" and include a referencedepends on the gene in which the change is found; DMD:c.[76A>C];GJB:c.[87delG].
- mosaics: two different nucleotides at the same position of the same allele are described in square brackets, separated by "/"; c.[=/83G>C].
- chimeras: two different nucleotides in the same position of the same allele are described in square brackets, separated by "//"; c.[=//83G>C].
Transcribed.
Secondly, we will have to look for the transcript in which the variant falls, since the position that identifies the nucleotide change is counted from the beginning of said transcript. This is necessary because most genes give rise to several different messenger RNA molecules with different lengths (transcripts), as an evolutionary mechanism to increase genetic diversity, which in turn will ultimately translate different isoforms of the protein that the gene encodes.
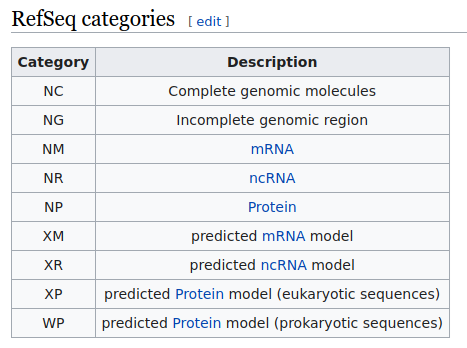
Given the large number of existing transcripts, it has been necessary to generate specific databases to identify them. The most used is RefSeq, which has indexed all naturally occurring human biological molecules based on nucleotides (DNA, RNA) and associated with a protein, encoded with a unique number for each record.
We will usually find the transcript code on which the position of the base change is based in an analysis table of the report of the detected variant, preceded by the letters NM (which refer to messenger RNA). There are other categories in RefSeq, such as NG or NC.
Reference genome.
Thirdly, we must know the reference genome that has been used for the analysis, since the advancement of scientific knowledge means that updates to said reference genome are being made. He reference genome consortium It is the entity in charge of maintaining, reviewing and accepting the modifications in the reference genome that are proposed by the international scientific community, updating successive versions of said reference genome. GRCh38 (Genome Reference Consortium human 38) is currently being used.
In successive versions, existing variants in different human populations have been incorporated, with the aim of representing the global genetic diversity as accurately as possible, which is why the project has progressively increased in complexity.
We will usually find this information in the appendices of the report, included in the chapter dedicated to methodology.