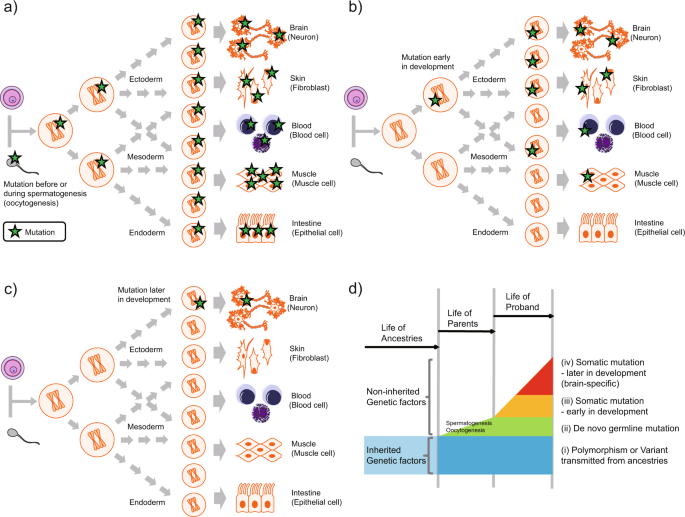
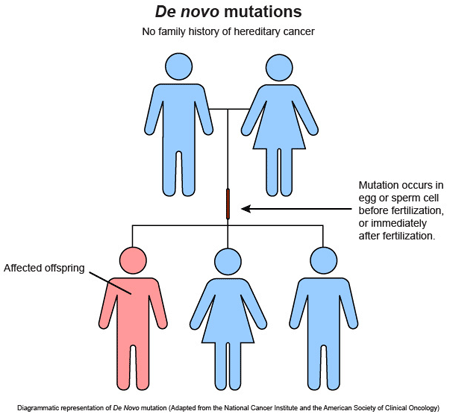
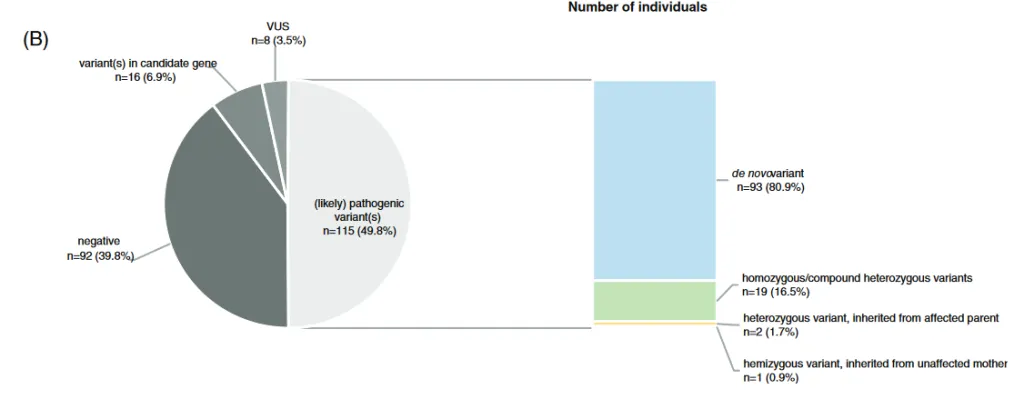
Durante los últimos años las mutaciones «de novo», aquellas no heredadas de ninguno de los dos progenitores, han cobrado especial relevancia en neurología pediátrica, por varios motivos:
Son responsables de la mayor parte de los casos de trastornos del neurodesarrollo identificables genéticamente.
Son responsables de los casos de mayor gravedad.
Esto se explica porque existe sesgo por selección natural. Las enfermedades heredadas suelen tener menor gravedad que las enfermedades «de novo», porque implican que el indivíduo ha sido capaz de vivir hasta la edad adulta, emparejase y reproducirse. De esta forma, las variantes que permanecen en la población general, y son encontradas con frecuencia en portadores sanos, producen menor repercusión fenotípica, aunque que una variante sea «de novo» no garantiza la patogenicidad.
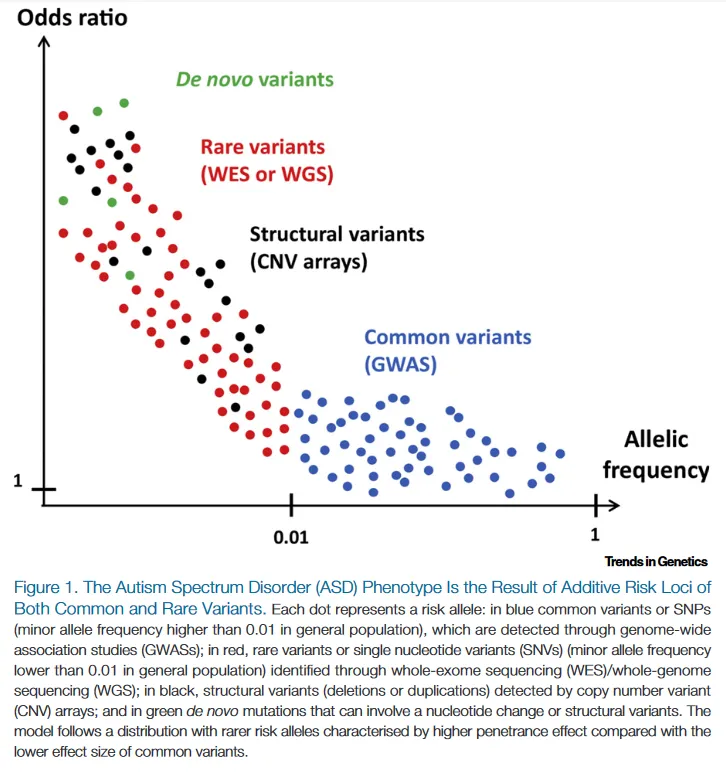
Cuestionan el paradigma clásico de enfermedad genética como enfermedad heredada.
Esto pone en cuestión la importancia relativa de los antecedentes familiares a la hora de sospechar una enfermedad genética, ya que su ausencia no es criterio de exclusión.
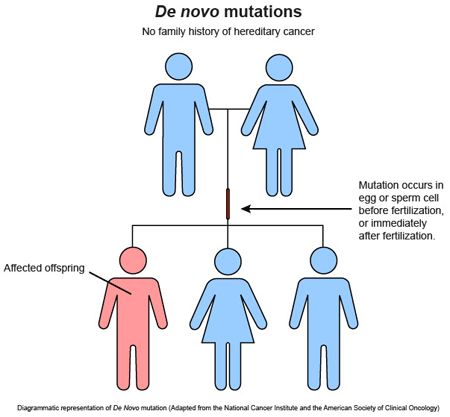
El riesgo de mutación de novo depende de la edad parental.
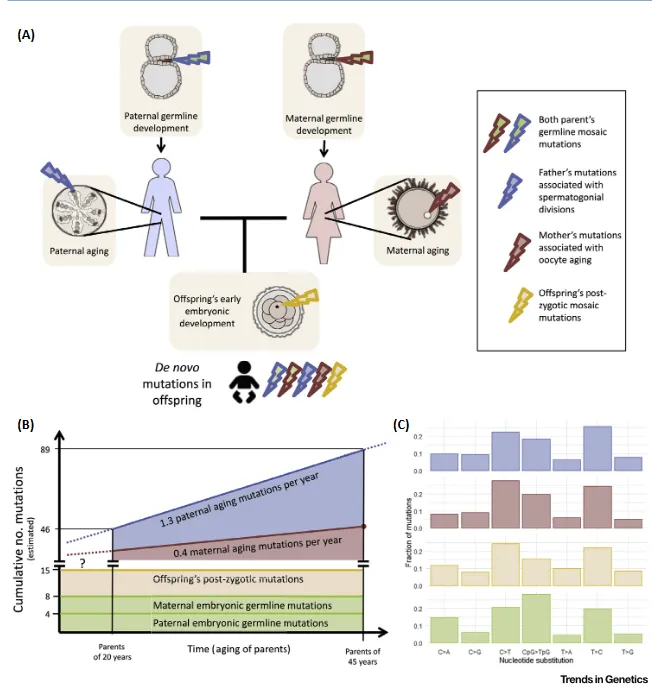
A través de los estudios genómicos, somos conocedores actualmente de que las personas sanas son portadoras de alrededor de unas 50-100 mutaciones de novo.
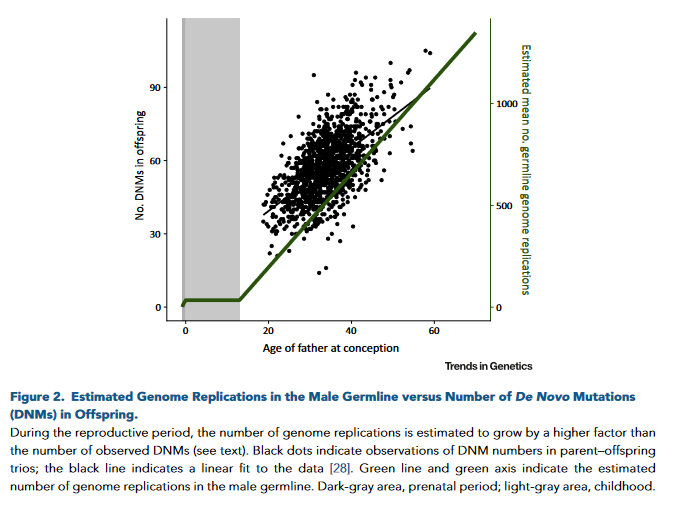
Por lo tanto, no es infrecuente que existan casos de fenotipo complejo en los que múltiples mutaciones dan lugar a fenotipos superpuestos de mayor gravedad (double trouble), dando lugar al concepto de «carga mutacional».
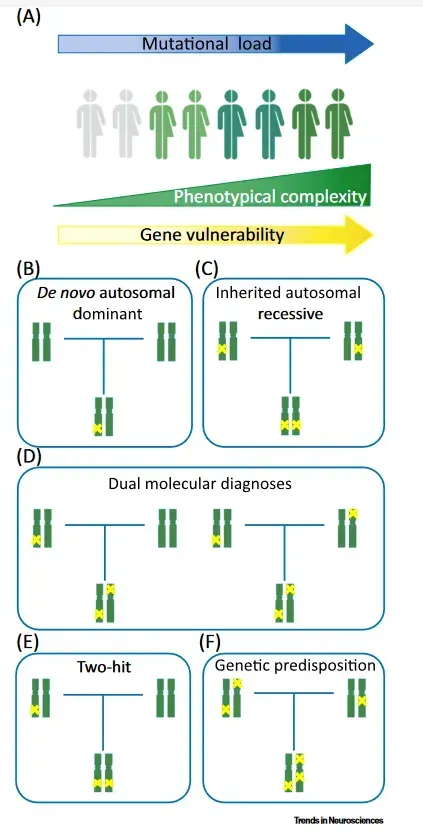
Esto cuestiona el paradigma clásico de enfermedad monogenética.
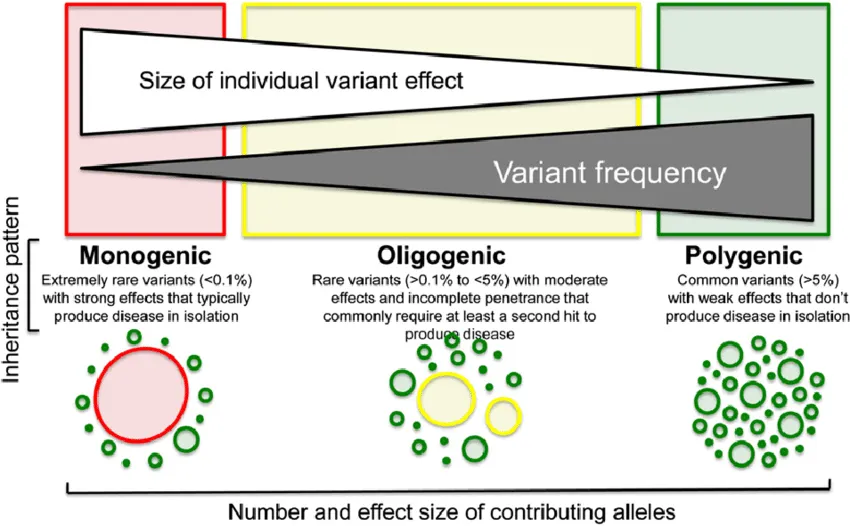
De esta forma, el papel de las variantes genéticas que cada indivíduo presenta puede representar una herencia poligénica (cuantitativa, factor de riesgo) cuando las variantes producen poca repercusión fenotípica, o una enfermedad monogenética, cuando la localización y consecuencias de la variante produce una repercusión clínica grave.
Pueden producirse más allá de la concepción, por lo que son la causa de los mosaicismos constitucionales.