
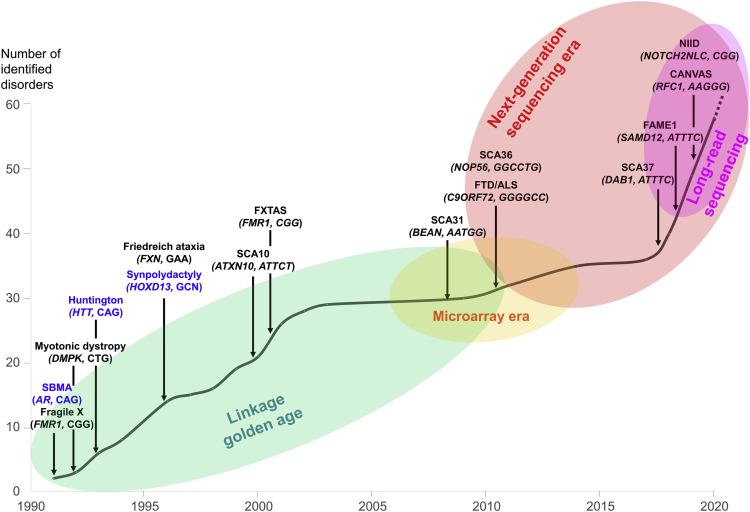
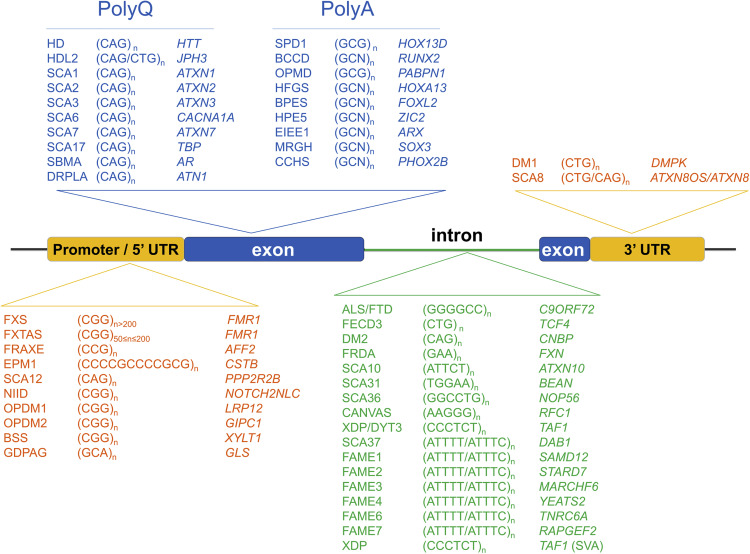
Enfermedades neurológicas causadas por STR.
| Abbreviated phenotype (MIM number) | Gene | Mode of inheritance | Repeat Motif | Location on Gene | Pathogenic repeat numbera | Chromosome | Coordinates (hg38) | Clinical phenotype | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| C9-FTD C9-ALS (#10550) | C9orf72 | AD | GGGGCC | 5’ Region | 24–4000 | chr9 | 27573485 | 27573546 | Frontotemporal dementia, amyotrophic lateral sclerosis | [32, 47, 65] |
| CANVAS (#614575) | RFC1 | AR | (AAGGG)400–2000 (ACAGG)exp AAAAG (normal) | Intron 2 | 400–2000 | chr4 | 39348425 | 39348483 | Cerebellar ataxia, neuropathy, and vestibular areflexia syndrome | [11, 28, 138] |
| DM1 (#160900) | DMPK | AD | CTG (Interruptions: CCG) | 3’ Region | 50–10,000 | chr19 | 45770205 | 45770266 | Myotonic dystrophy 1 | [60, 176] |
| DM2 (#602668) | CNBP (ZNF9) | AD | CCTG | Intron 1 | 50–11,000 | chr3 | 129172577 | 129172656 | Myotonic dystrophy 2 | [176] |
| DRPLA (#125370) | ATN1 | AD | CAG | Exon 5 | 49–93 | chr12 | 6936717 | 6936775 | Dentatorubral-pallidoluysian atrophy | [78] |
| EIEE1/XLID (#308350) (#300419) (#300215) | ARX | XL | GCC | Exon 2 | 17–27 | chrX | 25013654 | 25013697 | Clinical spectrum of disorders including developmental and epileptic encephalopathy 1, hydranencephaly with abnormal genitalia, X-linked lissencephaly 2 and X-linked mental retardation 29 | [73, 150] |
| FAME1 (#601068) | SAMD12 | AD | TTTCA within TTTTA repeat region | Intron 4 | 105–3680 | chr8 | 118366813 | 118366918 | Familial adult myoclonic epilepsy 1 | [22, 68] |
| FAME2 (#607876) | STARD7 | AD | ATTTC within ATTTT repeat region | Intron 1 | 150–460 | chr2 | 96197067 | 96197124 | Familial adult myoclonic epilepsy 2 | [27] |
| FAME3 (#613608) | MARCHF6 | AD | TTTCA within TTTTA repeat region | Intron 1 | 700–1035 | chr5 | 10356339 | 10356411 | Familial adult myoclonic epilepsy 3 | [40] |
| FAME6 (#618074) | TNRC6A | AD | TTTCA within TTTTA repeat region | Intron 1 | ? (only 1 family) | chr16 | 24613439 | 24613532 | Familial adult myoclonic epilepsy 6 | [68] |
| FAME7 (#618075) | RAPGEF2 | AD | TTTCA within TTTTA repeat region | Intron 14 | ? (only 1 family) | chr4 | 159342527 | 159342618 | Familial adult myoclonic epilepsy 7 | [68] |
| FRAXE (#309548) | FMR2 (AFF2) | XLR | CCG | 5’ Region | > 200 | chrX | 148500605 | 148500753 | Mental retardation, X-linked, FRAXE type | [53] |
| FRDA (#229300) | FXN | AR | GAA | Intron 1 | 66–1300 | chr9 | 69037275 | 69037314 | Friedreich ataxia | [5, 19, 162] |
| FXS (#300624) FXTAS (#300623) | FMR1 | XL | CGG | 5’ Region | 200–3000 55–200 | chrX | 147911979 | 147912111 | Fragile X syndrome Fragile X tremor/ataxia syndrome, premature ovarian failure 1 | [162] [56] |
| HD (#143100) | HTT | AD | CAG (Interruptions: CAA) | Exon 1 | 36–250 | chr4 | 3074876 | 3074941 | Huntington disease | [96, 101] |
| HDL1 (#603218) | PRNP | AD | 24-base octapeptide PHGGGWGQ | Exon 2 | 8–14 | chr20 | 4699379 | 4699380 | Huntington disease-like 1 | [108] |
| HDL2 (#606438) | JPH3 | AD | CTG | Exon 2A | 40–59 | chr16 | 87604283 | 87604329 | Huntington disease-like 2 | [62] |
| HMN | VWA1 | AR | GGCGCGGAGC | Exon 1 | 3 | chr1 | 1435799 | 1435820 | Hereditary axonal motor neuropathy | [121] |
| NIID (#603472) | NOTCH2NLC | AD | CGG | 5′ Region | 66–517 | chr1 | 149390803 | 149390842 | Neuronal intranuclear inclusion disease | [55, 118, 146] |
| OPDM1 (#164310) | LRP12 | AD | CGG | 5′ Region | 90–130 | chr8 | 104588965 | 104588999 | Oculopharyngodistal myopathy | [69] |
| OPDM2 (#618940) | GIPC1 | AD | CGG | 5’ Region | 70–164 | chr19 | 14496029 | 14496104 | Oculopharyngodistal myopathy | [172] |
| OPMD (#164300) | PABPN1 | AD | GCG | Exon 1 | 7–18 | chr14 | 23321472 | 23321511 | Oculopharyngeal muscular dystrophy | [15, 129] |
| OPML1 (#618637) | NUTM2B-AS1 | AD | CGG | 5′ Region | 16–160 | chr10 | 79826364 | 79826403 | Oculopharyngeal myopathy with leukoencephalopathy 1 | [69] |
| SBMA (#313200) | AR | XLR | CAG | Exon 1 | 38–68 | chrX | 67545317 | 67545419 | Spinal and bulbar muscular atrophy of Kennedy (Kennedy’s disease) | [44, 82, 147] |
| SCA1 (#164400) | ATXN1 | AD | CAG (Interruptions: CAT) | Exon 8 | 39–91 | chr6 | 16327636 | 16327723 | Spinocerebellar ataxia 1 | [120, 141] |
| SCA2 (#183090) | ATXN2 | AD | CAG (Interruptions: CAA, CGG, CGC) | Exon 1 | 33–200 (29–32 increased ALS risk) | chr12 | 111598950 | 111599019 | Spinocerebellar ataxia 2 | [18, 133, 141, 148] |
| SCA3 (#109150) | ATXN3 | AD | CAG | Exon 10 | 53–87 | chr14 | 92071011 | 92071052 | Spinocerebellar ataxia 3 | [74] |
| SCA6 (183086) | CACNA1A | AD | CAG | Exon 47 | 19–33 | chr19 | 13207858 | 13207897 | Spinocerebellar ataxia 6 | [141, 181] |
| SCA7 (#164500) | ATXN7 | AD | CAG | Exon 1 | 34–460 | chr3 | 63912685 | 63912716 | Spinocerebellar ataxia 7 | [18, 30] |
| SCA8 (#608768) | ATXN8 | AD | CAG/TAG | 3’ UTR | 74–1300 | chr13 | 70139383 | 70139428 | Spinocerebellar ataxia 8 | [79, 141, 155] |
| SCA10 (#603516) | ATXN10 | AD | ATTCT (Interruptions: ATCCT) | Intron 9 | 280–4500 | chr22 | 45795355 | 45795424 | Spinocerebellar ataxia 10 | [88, 100, 141] |
| SCA12 (#604326) | PPP2R2B | AD | CAG | 5’ Region | 51–78 | chr5 | 146878729 | 146878758 | Spinocerebellar ataxia 12 | [63, 94, 141] |
| SCA17 (#607136) | TBP | AD | CAG (Interruptions: CAT, CAA) | Exon 3 | 43–66 | chr6 | 170561907 | 170562017 | Spinocerebellar ataxia 17, Huntington disease-like 4 | [97, 115, 141] |
| SCA31 (#117210) | BEAN1 | AD | TGGAA within TAAAA and TAGAA repeat region | Intron/ Intergenic region | 500–760 (> 110 TGGAA repeats) | chr16 | 66495475 | 66495509 | Spinocerebellar ataxia 31 | [134] |
| SCA36 (#614153) | NOP56 | AD | GGCCTG | Intron 1 | 650–2500 | chr20 | 2652733 | 2652775 | Spinocerebellar ataxia 36 | [77] |
| SCA37 (#615945) | DAB1 | AD | ATTTC within (ATTTT)7–400 repeat region | 5’ Region | 31–75 | chr1 | 57367044 | 57367125 | Spinocerebellar ataxia 37 | [139] |
| ULD (#254800) | CSTB | AR | CCCCGCCCCGCG | Upstream 5’ UTR | 30–125 | chr21 | 43776444 | 43776479 | Progressive myoclonic epilepsy 1A (Unverricht and Lundborg disease) | [87, 91] |
aThese ranges vary between studies and often the upper limit is unknown. It is important to note that these are only potentially pathogenic. There is a small (< 1%) subsection of the healthy control population who have expanded alleles with no clinical manifestations. Similarly, there are alleles lower than the given range who may have intermediate alleles and premutation syndromes
Enfermedades congénitas y del desarrollo causadas por STR.
| Phenotype (OMIM #) | Gene | Motif | Pathogenic repeat number | Location | (hg38) | References | ||
|---|---|---|---|---|---|---|---|---|
| BPES (#110100) | FOXL2 | GCG | 22–24 | Exon | chr3 | 138946022 | 138946062 | [116] |
| CCHS (#209880) | PHOX2B | GCG | 24–33 | Exon | chr4 | 41745976 | 41746022 | [7] |
| DBQD2 (#615777) | XYLT1 | GGC | 100–800 | 5’ Region | chr16 | 17470869 | 17470967 | [86] |
| FECD3 (#613267) | TCF4 | TGC | > 50 | Intron | chr18a | 55222184a | 55635956a | [167] |
| GDPAG (#618412) | GLS | GCA | > 300 | 5’ Region | chr2 | 190880873 | 190880920 | [159] |
| HFG (#140000) | HOXA13 | GCG | 24–26 | Exon | chr7 | 27199827 | 27199967 | [50] |
| HPE5 (#609637) | ZIC2 | GCG | 25 | Exon | chr13 | 99985449 | 99985494 | [17] |
| HSAN8 (#616488) | PRDM12 | GCG | 18–19 | Exon | chr9 | 130681606 | 130681641 | [23] |
| SPD1 (#186000) | HOXD13 | GCG | 22–29 | Exon | chr2 | 176093058 | 176093099 | [2] |
| XLMR (#300123) | SOX3 | GCG | 15–26 | Exon | chr3 | 181712415 | 181712456 | [89] |
aLocation of entire gene listed
332710
{332710:XQQ8IRAK},{332710:6A56W7C4}
1
vancouver
50
default
2455
https://neuropediatoolkit.org/wp-content/plugins/zotpress/
%7B%22status%22%3A%22success%22%2C%22updateneeded%22%3Afalse%2C%22instance%22%3Afalse%2C%22meta%22%3A%7B%22request_last%22%3A0%2C%22request_next%22%3A0%2C%22used_cache%22%3Atrue%7D%2C%22data%22%3A%5B%7B%22key%22%3A%22XQQ8IRAK%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Depienne%20and%20Mandel%22%2C%22parsedDate%22%3A%222021-05-06%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BDepienne%20C%2C%20Mandel%20JL.%2030%20years%20of%20repeat%20expansion%20disorders%3A%20What%20have%20we%20learned%20and%20what%20are%20the%20remaining%20challenges%3F%20The%20American%20Journal%20of%20Human%20Genetics%20%5BInternet%5D.%202021%20May%206%20%5Bcited%202021%20Sept%2019%5D%3B108%285%29%3A764%26%23x2013%3B85.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0002929721000951%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0002929721000951%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%2230%20years%20of%20repeat%20expansion%20disorders%3A%20What%20have%20we%20learned%20and%20what%20are%20the%20remaining%20challenges%3F%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christel%22%2C%22lastName%22%3A%22Depienne%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jean-Louis%22%2C%22lastName%22%3A%22Mandel%22%7D%5D%2C%22abstractNote%22%3A%22Tandem%20repeats%20represent%20one%20of%20the%20most%20abundant%20class%20of%20variations%20in%20human%20genomes%2C%20which%20are%20polymorphic%20by%20nature%20and%20become%20highly%20unstable%20in%20a%20length-dependent%20manner.%20The%20expansion%20of%20repeat%20length%20across%20generations%20is%20a%20well-established%20process%20that%20results%20in%20human%20disorders%20mainly%20affecting%20the%20central%20nervous%20system.%20At%20least%2050%20disorders%20associated%20with%20expansion%20loci%20have%20been%20described%20to%20date%2C%20with%20half%20recognized%20only%20in%20the%20last%20ten%20years%2C%20as%20prior%20methodological%20difficulties%20limited%20their%20identification.%20These%20limitations%20still%20apply%20to%20the%20current%20widely%20used%20molecular%20diagnostic%20methods%20%28exome%20or%20gene%20panels%29%20and%20thus%20result%20in%20missed%20diagnosis%20detrimental%20to%20affected%20individuals%20and%20their%20families%2C%20especially%20for%20disorders%20that%20are%20very%20rare%20and%5C%2For%20clinically%20not%20recognizable.%20Most%20of%20these%20disorders%20have%20been%20identified%20through%20family-driven%20approaches%20and%20many%20others%20likely%20remain%20to%20be%20identified.%20The%20recent%20development%20of%20long-read%20technologies%20provides%20a%20unique%20opportunity%20to%20systematically%20investigate%20the%20contribution%20of%20tandem%20repeats%20and%20repeat%20expansions%20to%20the%20genetic%20architecture%20of%20human%20disorders.%20In%20this%20review%2C%20we%20summarize%20the%20current%20and%20most%20recent%20knowledge%20about%20the%20genetics%20of%20repeat%20expansion%20disorders%20and%20the%20diversity%20of%20their%20pathophysiological%20mechanisms%20and%20outline%20the%20perspectives%20of%20developing%20personalized%20treatments%20in%20the%20future.%22%2C%22date%22%3A%22May%206%2C%202021%22%2C%22section%22%3A%22%22%2C%22partNumber%22%3A%22%22%2C%22partTitle%22%3A%22%22%2C%22DOI%22%3A%2210.1016%5C%2Fj.ajhg.2021.03.011%22%2C%22citationKey%22%3A%22%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0002929721000951%22%2C%22PMID%22%3A%22%22%2C%22PMCID%22%3A%22%22%2C%22ISSN%22%3A%220002-9297%22%2C%22language%22%3A%22en%22%2C%22collections%22%3A%5B%22W9TVQG8W%22%5D%2C%22dateModified%22%3A%222025-11-10T06%3A48%3A12Z%22%7D%7D%2C%7B%22key%22%3A%226A56W7C4%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Chintalaphani%20et%20al.%22%2C%22parsedDate%22%3A%222021-05-25%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BChintalaphani%20SR%2C%20Pineda%20SS%2C%20Deveson%20IW%2C%20Kumar%20KR.%20An%20update%20on%20the%20neurological%20short%20tandem%20repeat%20expansion%20disorders%20and%20the%20emergence%20of%20long-read%20sequencing%20diagnostics.%20Acta%20Neuropathologica%20Communications%20%5BInternet%5D.%202021%20May%2025%20%5Bcited%202021%20Sept%2019%5D%3B9%281%29%3A98.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1186%5C%2Fs40478-021-01201-x%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1186%5C%2Fs40478-021-01201-x%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22An%20update%20on%20the%20neurological%20short%20tandem%20repeat%20expansion%20disorders%20and%20the%20emergence%20of%20long-read%20sequencing%20diagnostics%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sanjog%20R.%22%2C%22lastName%22%3A%22Chintalaphani%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sandy%20S.%22%2C%22lastName%22%3A%22Pineda%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ira%20W.%22%2C%22lastName%22%3A%22Deveson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Kishore%20R.%22%2C%22lastName%22%3A%22Kumar%22%7D%5D%2C%22abstractNote%22%3A%22Short%20tandem%20repeat%20%28STR%29%20expansion%20disorders%20are%20an%20important%20cause%20of%20human%20neurological%20disease.%20They%20have%20an%20established%20role%20in%20more%20than%2040%20different%20phenotypes%20including%20the%20myotonic%20dystrophies%2C%20Fragile%20X%20syndrome%2C%20Huntington%5Cu2019s%20disease%2C%20the%20hereditary%20cerebellar%20ataxias%2C%20amyotrophic%20lateral%20sclerosis%20and%20frontotemporal%20dementia.%22%2C%22date%22%3A%22May%2025%2C%202021%22%2C%22section%22%3A%22%22%2C%22partNumber%22%3A%22%22%2C%22partTitle%22%3A%22%22%2C%22DOI%22%3A%2210.1186%5C%2Fs40478-021-01201-x%22%2C%22citationKey%22%3A%22%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1186%5C%2Fs40478-021-01201-x%22%2C%22PMID%22%3A%22%22%2C%22PMCID%22%3A%22%22%2C%22ISSN%22%3A%222051-5960%22%2C%22language%22%3A%22%22%2C%22collections%22%3A%5B%22W9TVQG8W%22%5D%2C%22dateModified%22%3A%222025-04-22T08%3A25%3A17Z%22%7D%7D%5D%7D
1.
Depienne C, Mandel JL. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? The American Journal of Human Genetics [Internet]. 2021 May 6 [cited 2021 Sept 19];108(5):764–85. Available from: https://www.sciencedirect.com/science/article/pii/S0002929721000951
1.
Chintalaphani SR, Pineda SS, Deveson IW, Kumar KR. An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathologica Communications [Internet]. 2021 May 25 [cited 2021 Sept 19];9(1):98. Available from: https://doi.org/10.1186/s40478-021-01201-x