Las enfermedades mitocondriales son un grupo de trastornos genéticos que afectan las mitocondrias, las estructuras celulares encargadas de producir energía en forma de ATP (adenosín trifosfato). Estas enfermedades son causadas por mutaciones en el ADN mitocondrial o en los genes nucleares que codifican proteínas relacionadas con la función mitocondrial. Debido a que las mitocondrias son cruciales para el metabolismo energético de las células, los trastornos mitocondriales pueden afectar múltiples sistemas del cuerpo, incluyendo el cerebro, el sistema nervioso, los músculos, el corazón y otros órganos.
Características o peculiaridades de las enfermedades mitocondriales.
| ADN Nuclear | ADNmt | |
| Localización | Nucleo | Matriz mitocondrial |
| Tamaño | 3200 Mb | 16.6 kb |
| Estructura | Doble hélice antiparalela linear | Doble hélice circular |
| Intrones | Presentes | Ausentes |
| Secuencias no codificantes | El 98% | El 7% |
| Transcripción | Monogénica. Tránscritos individuales para cada gen. | Multigénica. Transcripción en bloque de todo el tránscrito. |
| Número de copias | 1 set por célula | De cientos a miles por celula |
| Proteinas asociadas | Histonas | Sin proteinas asociadas |
| Genes codificantes | 30.000 | 37 |
| Codon | Código genético universal | Código genético mitocondrial |
- Heteroplasmia. La heteroplasmia se refiere a la presencia de diferentes tipos de ADN mitocondrial en una célula o individuo. Las mitocondrias se multiplican dividiéndose de forma independiente al ciclo celular, y en el proceso de copiado pueden producirse errores que se van acumulando con el tiempo. Esta variación en el ADN mitocondrial se conoce como heteroplasmia.
- Herencia mitocondrial. Las mitocondrias se heredan principalmente a través de la madre, y por lo tanto, el ADN mitocondrial (ADNmt) se hereda exclusivamente de la madre, a diferencia del ADN nuclear, que se hereda de ambos progenitores. Las mutaciones o variantes en el ADN mitocondrial se transmiten de madre a hijos, pero solo las hijas pueden transmitir estas mutaciones a sus descendientes. Los hijos varones no transmiten su ADN mitocondrial a su descendencia.
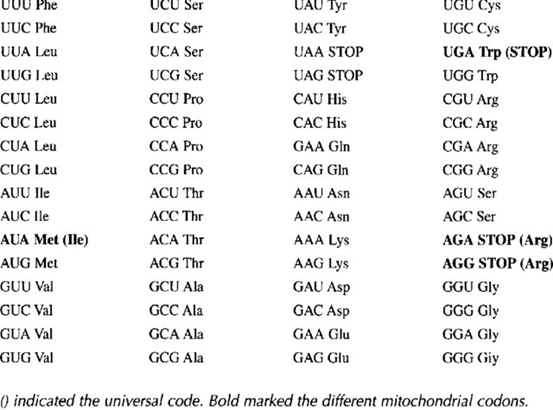
- Código genético mitocondrial. El código genético mitocondrial es el conjunto de reglas que determina cómo la información contenida en el ADN mitocondrial (ADNmt) se traduce en proteínas dentro de las mitocondrias, y es similar pero no idéntico al código genético universal utilizado en el resto de la célula, que se encuentra en el ADN nuclear. En el código genético mitocondrial, algunas de las codificaciones de aminoácidos son diferentes, lo que significa que ciertos codones en el ARNm se traducen en aminoácidos diferentes en comparación con el código genético nuclear. Algunas de las diferencias más notables en el código genético mitocondrial son:
- El codón AGA en el código nuclear codifica para el aminoácido arginina, mientras que en el código mitocondrial codifica para la serina.
- El codón AGG en el código nuclear codifica para el aminoácido arginina, mientras que en el código mitocondrial también codifica para la serina.
- El codón UGA en el código nuclear suele ser un codón de parada (codón de terminación), pero en el código mitocondrial codifica para el aminoácido triptófano.
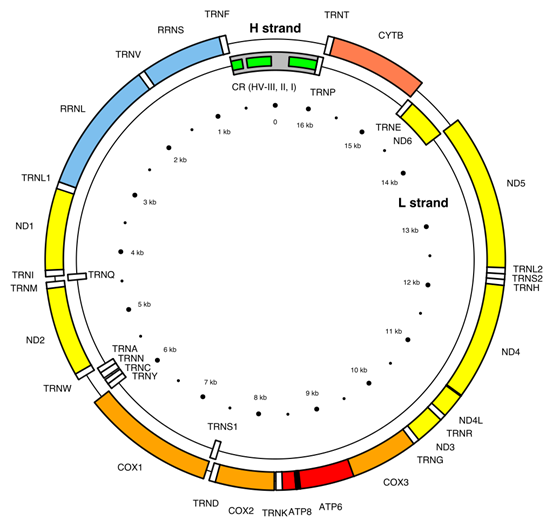
- Cromosoma mitocondrial. El término «cromosoma mitocondrial» se refiere al material genético contenido en las mitocondrias, pero es importante destacar que las mitocondrias no tienen estructuras cromosómicas similares a los cromosomas del núcleo. Aquí hay algunas características clave del ADN mitocondrial:
- Forma y Estructura: El ADN mitocondrial suele presentarse en forma de un círculo cerrado, en contraste con los cromosomas lineales en el núcleo.
- Genes: El ADN mitocondrial contiene un total de 37 genes.
- Transcripción y Traducción: Las mitocondrias son capaces de transcribir y traducir su propio ADN, lo que les permite sintetizar las proteínas necesarias para su función. Sin embargo, la mayoría de las proteínas requeridas para la función mitocondrial son codificadas por el núcleo y luego importadas a las mitocondrias.
Genes codificados en el ADNmt
The 37 genes of the Cambridge Reference Sequence for human mitochondrial DNA and their locations[30]
| Gene | Type | Product | Positions in the mitogenome | Strand |
| MT-ATP8 | protein coding | ATP synthase, Fo subunit 8 (complex V) | 08,366–08,572 (overlap with MT-ATP6) | H |
| MT-ATP6 | protein coding | ATP synthase, Fo subunit 6 (complex V) | 08,527–09,207 (overlap with MT-ATP8) | H |
| MT-CO1 | protein coding | Cytochrome c oxidase, subunit 1 (complex IV) | 05,904–07,445 | H |
| MT-CO2 | protein coding | Cytochrome c oxidase, subunit 2 (complex IV) | 07,586–08,269 | H |
| MT-CO3 | protein coding | Cytochrome c oxidase, subunit 3 (complex IV) | 09,207–09,990 | H |
| MT-CYB | protein coding | Cytochrome b (complex III) | 14,747–15,887 | H |
| MT-ND1 | protein coding | NADH dehydrogenase, subunit 1 (complex I) | 03,307–04,262 | H |
| MT-ND2 | protein coding | NADH dehydrogenase, subunit 2 (complex I) | 04,470–05,511 | H |
| MT-ND3 | protein coding | NADH dehydrogenase, subunit 3 (complex I) | 10,059–10,404 | H |
| MT-ND4L | protein coding | NADH dehydrogenase, subunit 4L (complex I) | 10,470–10,766 (overlap with MT-ND4) | H |
| MT-ND4 | protein coding | NADH dehydrogenase, subunit 4 (complex I) | 10,760–12,137 (overlap with MT-ND4L) | H |
| MT-ND5 | protein coding | NADH dehydrogenase, subunit 5 (complex I) | 12,337–14,148 | H |
| MT-ND6 | protein coding | NADH dehydrogenase, subunit 6 (complex I) | 14,149–14,673 | L |
| MT-RNR2 | protein coding | Humanin | — | — |
| MT-TA | transfer RNA | tRNA-Alanine (Ala or A) | 05,587–05,655 | L |
| MT-TR | transfer RNA | tRNA-Arginine (Arg or R) | 10,405–10,469 | H |
| MT-TN | transfer RNA | tRNA-Asparagine (Asn or N) | 05,657–05,729 | L |
| MT-TD | transfer RNA | tRNA-Aspartic acid (Asp or D) | 07,518–07,585 | H |
| MT-TC | transfer RNA | tRNA-Cysteine (Cys or C) | 05,761–05,826 | L |
| MT-TE | transfer RNA | tRNA-Glutamic acid (Glu or E) | 14,674–14,742 | L |
| MT-TQ | transfer RNA | tRNA-Glutamine (Gln or Q) | 04,329–04,400 | L |
| MT-TG | transfer RNA | tRNA-Glycine (Gly or G) | 09,991–10,058 | H |
| MT-TH | transfer RNA | tRNA-Histidine (His or H) | 12,138–12,206 | H |
| MT-TI | transfer RNA | tRNA-Isoleucine (Ile or I) | 04,263–04,331 | H |
| MT-TL1 | transfer RNA | tRNA-Leucine (Leu-UUR or L) | 03,230–03,304 | H |
| MT-TL2 | transfer RNA | tRNA-Leucine (Leu-CUN or L) | 12,266–12,336 | H |
| MT-TK | transfer RNA | tRNA-Lysine (Lys or K) | 08,295–08,364 | H |
| MT-TM | transfer RNA | tRNA-Methionine (Met or M) | 04,402–04,469 | H |
| MT-TF | transfer RNA | tRNA-Phenylalanine (Phe or F) | 00,577–00,647 | H |
| MT-TP | transfer RNA | tRNA-Proline (Pro or P) | 15,956–16,023 | L |
| MT-TS1 | transfer RNA | tRNA-Serine (Ser-UCN or S) | 07,446–07,514 | L |
| MT-TS2 | transfer RNA | tRNA-Serine (Ser-AGY or S) | 12,207–12,265 | H |
| MT-TT | transfer RNA | tRNA-Threonine (Thr or T) | 15,888–15,953 | H |
| MT-TW | transfer RNA | tRNA-Tryptophan (Trp or W) | 05,512–05,579 | H |
| MT-TY | transfer RNA | tRNA-Tyrosine (Tyr or Y) | 05,826–05,891 | L |
| MT-TV | transfer RNA | tRNA-Valine (Val or V) | 01,602–01,670 | H |
| MT-RNR1 | ribosomal RNA | Small subunit : SSU (12S) | 00,648–01,601 | H |
| MT-RNR2 | ribosomal RNA | Large subunit : LSU (16S) | 01,671–03,229 | H |
Criterios diagnósticos de enfermedad mitocondrial. Cuando sospechar la presencia de una enfermedad mitocondrial.
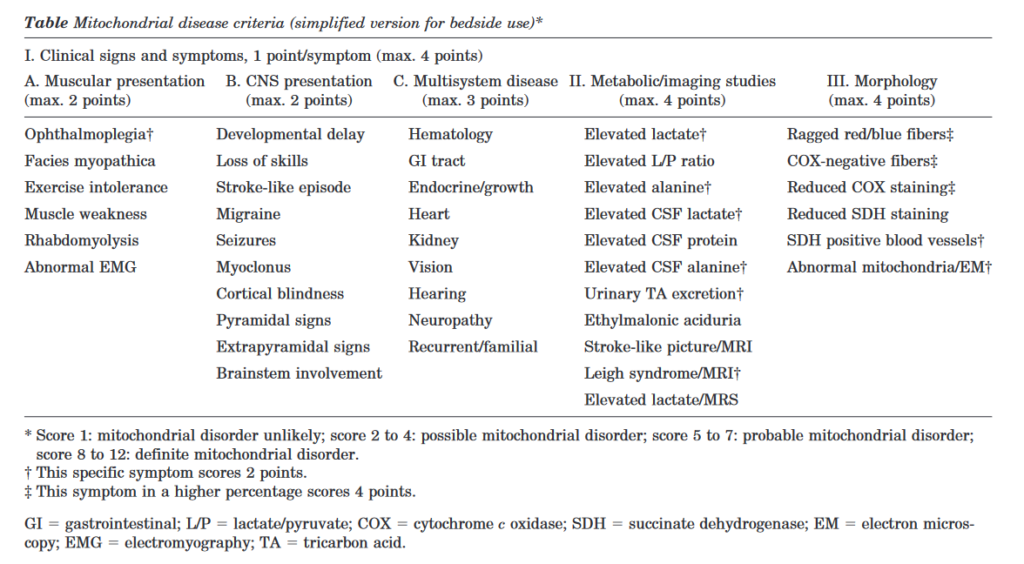
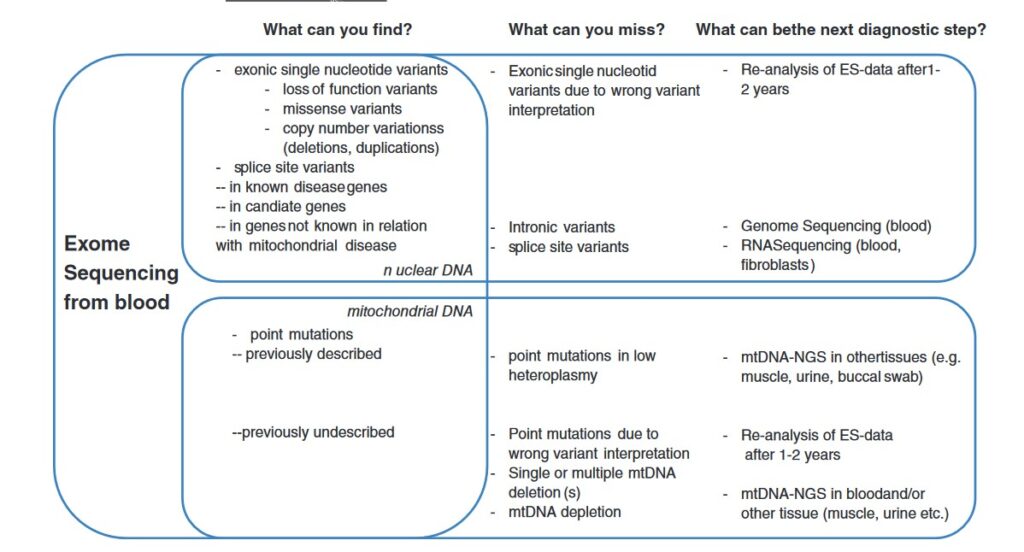
Cuando solicitar estudio de enfermedades mitocodriales si se ha realizado secuenciación NGS. Que te puedes perder si has realizado una secuenciación NGS y buscas una enfermedad mitocondrial.
{332710:BD8ICUPB};{332710:JZJ6PPCJ}
vancouver
default
asc
0
6412
%7B%22status%22%3A%22success%22%2C%22updateneeded%22%3Afalse%2C%22instance%22%3Afalse%2C%22meta%22%3A%7B%22request_last%22%3A5750%2C%22request_next%22%3A50%2C%22used_cache%22%3Atrue%7D%2C%22data%22%3A%5B%7B%22key%22%3A%226DUU5I4T%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Specchio%20et%20al.%22%2C%22parsedDate%22%3A%222022%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BSpecchio%20N%2C%20Wirrell%20EC%2C%20Scheffer%20IE%2C%20Nabbout%20R%2C%20Riney%20K%2C%20Samia%20P%2C%20et%20al.%20International%20League%20Against%20Epilepsy%20classification%20and%20definition%20of%20epilepsy%20syndromes%20with%20onset%20in%20childhood%3A%20Position%20paper%20by%20the%20ILAE%20Task%20Force%20on%20Nosology%20and%20Definitions.%20Epilepsia%20%5BInternet%5D.%202022%20%5Bcited%202022%20Oct%2022%5D%3B63%286%29%3A1398%26%23x2013%3B442.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1111%5C%2Fepi.17241%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1111%5C%2Fepi.17241%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22International%20League%20Against%20Epilepsy%20classification%20and%20definition%20of%20epilepsy%20syndromes%20with%20onset%20in%20childhood%3A%20Position%20paper%20by%20the%20ILAE%20Task%20Force%20on%20Nosology%20and%20Definitions%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nicola%22%2C%22lastName%22%3A%22Specchio%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Elaine%20C.%22%2C%22lastName%22%3A%22Wirrell%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ingrid%20E.%22%2C%22lastName%22%3A%22Scheffer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rima%22%2C%22lastName%22%3A%22Nabbout%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Kate%22%2C%22lastName%22%3A%22Riney%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pauline%22%2C%22lastName%22%3A%22Samia%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Marilisa%22%2C%22lastName%22%3A%22Guerreiro%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sam%22%2C%22lastName%22%3A%22Gwer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sameer%20M.%22%2C%22lastName%22%3A%22Zuberi%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jo%20M.%22%2C%22lastName%22%3A%22Wilmshurst%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Elissa%22%2C%22lastName%22%3A%22Yozawitz%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ronit%22%2C%22lastName%22%3A%22Pressler%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Edouard%22%2C%22lastName%22%3A%22Hirsch%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Samuel%22%2C%22lastName%22%3A%22Wiebe%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Helen%20J.%22%2C%22lastName%22%3A%22Cross%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Emilio%22%2C%22lastName%22%3A%22Perucca%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Solomon%20L.%22%2C%22lastName%22%3A%22Mosh%5Cu00e9%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Paolo%22%2C%22lastName%22%3A%22Tinuper%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22St%5Cu00e9phane%22%2C%22lastName%22%3A%22Auvin%22%7D%5D%2C%22abstractNote%22%3A%22The%202017%20International%20League%20Against%20Epilepsy%20classification%20has%20defined%20a%20three-tier%20system%20with%20epilepsy%20syndrome%20identification%20at%20the%20third%20level.%20Although%20a%20syndrome%20cannot%20be%20determined%20in%20all%20children%20with%20epilepsy%2C%20identification%20of%20a%20specific%20syndrome%20provides%20guidance%20on%20management%20and%20prognosis.%20In%20this%20paper%2C%20we%20describe%20the%20childhood%20onset%20epilepsy%20syndromes%2C%20most%20of%20which%20have%20both%20mandatory%20seizure%20type%28s%29%20and%20interictal%20electroencephalographic%20%28EEG%29%20features.%20Based%20on%20the%202017%20Classification%20of%20Seizures%20and%20Epilepsies%2C%20some%20syndrome%20names%20have%20been%20updated%20using%20terms%20directly%20describing%20the%20seizure%20semiology.%20Epilepsy%20syndromes%20beginning%20in%20childhood%20have%20been%20divided%20into%20three%20categories%3A%20%281%29%20self-limited%20focal%20epilepsies%2C%20comprising%20four%20syndromes%3A%20self-limited%20epilepsy%20with%20centrotemporal%20spikes%2C%20self-limited%20epilepsy%20with%20autonomic%20seizures%2C%20childhood%20occipital%20visual%20epilepsy%2C%20and%20photosensitive%20occipital%20lobe%20epilepsy%3B%20%282%29%20generalized%20epilepsies%2C%20comprising%20three%20syndromes%3A%20childhood%20absence%20epilepsy%2C%20epilepsy%20with%20myoclonic%20absence%2C%20and%20epilepsy%20with%20eyelid%20myoclonia%3B%20and%20%283%29%20developmental%20and%5C%2For%20epileptic%20encephalopathies%2C%20comprising%20five%20syndromes%3A%20epilepsy%20with%20myoclonic%5Cu2013atonic%20seizures%2C%20Lennox%5Cu2013Gastaut%20syndrome%2C%20developmental%20and%5C%2For%20epileptic%20encephalopathy%20with%20spike-and-wave%20activation%20in%20sleep%2C%20hemiconvulsion%5Cu2013hemiplegia%5Cu2013epilepsy%20syndrome%2C%20and%20febrile%20infection-related%20epilepsy%20syndrome.%20We%20define%20each%2C%20highlighting%20the%20mandatory%20seizure%28s%29%2C%20EEG%20features%2C%20phenotypic%20variations%2C%20and%20findings%20from%20key%20investigations.%22%2C%22date%22%3A%222022%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1111%5C%2Fepi.17241%22%2C%22ISSN%22%3A%221528-1167%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1111%5C%2Fepi.17241%22%2C%22collections%22%3A%5B%22IVUN2VXI%22%5D%2C%22dateModified%22%3A%222022-10-22T08%3A36%3A13Z%22%7D%7D%2C%7B%22key%22%3A%22MH735Z3S%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Kang%20and%20Kwon%22%2C%22parsedDate%22%3A%222014%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKang%20B%2C%20Kwon%20YS.%20Benign%20convulsion%20with%20mild%20gastroenteritis.%20Korean%20J%20Pediatr%20%5BInternet%5D.%202014%20%5Bcited%202022%20Oct%2022%5D%3B57%287%29%3A304.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.e-cep.org%5C%2Fjournal%5C%2Fview.php%3Fdoi%3D10.3345%5C%2Fkjp.2014.57.7.304%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.e-cep.org%5C%2Fjournal%5C%2Fview.php%3Fdoi%3D10.3345%5C%2Fkjp.2014.57.7.304%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Benign%20convulsion%20with%20mild%20gastroenteritis%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ben%22%2C%22lastName%22%3A%22Kang%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Young%20Se%22%2C%22lastName%22%3A%22Kwon%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222014%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.3345%5C%2Fkjp.2014.57.7.304%22%2C%22ISSN%22%3A%221738-1061%2C%202092-7258%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.e-cep.org%5C%2Fjournal%5C%2Fview.php%3Fdoi%3D10.3345%5C%2Fkjp.2014.57.7.304%22%2C%22collections%22%3A%5B%22EJJIHH7N%22%5D%2C%22dateModified%22%3A%222022-10-22T07%3A58%3A41Z%22%7D%7D%2C%7B%22key%22%3A%22QECZT2KY%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Lee%20et%20al.%22%2C%22parsedDate%22%3A%222021-12-27%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BLee%20YS%2C%20Lee%20GH%2C%20Kwon%20YS.%20Update%20on%20benign%20convulsions%20with%20mild%20gastroenteritis.%20Clin%20Exp%20Pediatr%20%5BInternet%5D.%202021%20Dec%2027%20%5Bcited%202022%20Oct%2022%5D%3B65%2810%29%3A469%26%23x2013%3B75.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.e-cep.org%5C%2Fjournal%5C%2Fview.php%3Fdoi%3D10.3345%5C%2Fcep.2021.00997%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.e-cep.org%5C%2Fjournal%5C%2Fview.php%3Fdoi%3D10.3345%5C%2Fcep.2021.00997%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Update%20on%20benign%20convulsions%20with%20mild%20gastroenteritis%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Yeong%20Seok%22%2C%22lastName%22%3A%22Lee%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ga%20Hee%22%2C%22lastName%22%3A%22Lee%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Young%20Se%22%2C%22lastName%22%3A%22Kwon%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222021%5C%2F12%5C%2F27%22%2C%22language%22%3A%22English%22%2C%22DOI%22%3A%2210.3345%5C%2Fcep.2021.00997%22%2C%22ISSN%22%3A%222713-4148%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.e-cep.org%5C%2Fjournal%5C%2Fview.php%3Fdoi%3D10.3345%5C%2Fcep.2021.00997%22%2C%22collections%22%3A%5B%22EJJIHH7N%22%5D%2C%22dateModified%22%3A%222022-10-22T07%3A58%3A20Z%22%7D%7D%2C%7B%22key%22%3A%22KKNL45RC%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Verity%20et%20al.%22%2C%22parsedDate%22%3A%222021-03%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BVerity%20C%2C%20Baker%20E%2C%20Maunder%20P%2C%20Pal%20S%2C%20Winstone%20AM.%20Differential%20diagnosis%20of%20progressive%20intellectual%20and%20neurological%20deterioration%20in%20children.%20Dev%20Med%20Child%20Neurol%20%5BInternet%5D.%202021%20Mar%20%5Bcited%202022%20Oct%2015%5D%3B63%283%29%3A287%26%23x2013%3B94.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC7891454%5C%2F%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC7891454%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Differential%20diagnosis%20of%20progressive%20intellectual%20and%20neurological%20deterioration%20in%20children%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christopher%22%2C%22lastName%22%3A%22Verity%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Elaine%22%2C%22lastName%22%3A%22Baker%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Polly%22%2C%22lastName%22%3A%22Maunder%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Suvankar%22%2C%22lastName%22%3A%22Pal%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Anne%20Marie%22%2C%22lastName%22%3A%22Winstone%22%7D%5D%2C%22abstractNote%22%3A%22The%20prevalence%20of%20diseases%20causing%20childhood%20progressive%20intellectual%20and%20neurological%20deterioration%20in%20the%20UK%20is%20low%20%28approximately%200.1%5C%2F1000%20live%20births%29.There%20were%20more%20than%20220%20different%20disorders%2C%20mainly%20genetically%20determined.The%20majority%20of%20disorders%20presented%20early%20in%20childhood%3A%2081%25%20before%20the%20age%20of%205%5Cu00a0years.There%20were%20similarities%20in%20the%20disease%20spectrum%20in%20White%20and%20Pakistani%20children.%5Cn%2C%20Video%20Podcast%3A%20https%3A%5C%2F%5C%2Fyoutu.be%5C%2FQbUG-ZVJLHc%5Cn%2C%20This%20article%20is%20commented%20on%20by%20van%20Karnebeek%20on%20page%20243%20of%20this%20issue.%22%2C%22date%22%3A%222021-3%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1111%5C%2Fdmcn.14691%22%2C%22ISSN%22%3A%220012-1622%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC7891454%5C%2F%22%2C%22collections%22%3A%5B%22I83TRK6C%22%5D%2C%22dateModified%22%3A%222022-10-15T17%3A49%3A16Z%22%7D%7D%2C%7B%22key%22%3A%22D36HAPFF%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Hoffmann%20et%20al.%22%2C%22parsedDate%22%3A%222006-08-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BHoffmann%20K%2C%20M%26%23xFC%3Bller%20JS%2C%20Stricker%20S%2C%20Megarbane%20A%2C%20Rajab%20A%2C%20Lindner%20TH%2C%20et%20al.%20Escobar%20Syndrome%20Is%20a%20Prenatal%20Myasthenia%20Caused%20by%20Disruption%20of%20the%20Acetylcholine%20Receptor%20Fetal%20%26%23x3B3%3B%20Subunit.%20The%20American%20Journal%20of%20Human%20Genetics%20%5BInternet%5D.%202006%20Aug%201%20%5Bcited%202022%20Oct%2015%5D%3B79%282%29%3A303%26%23x2013%3B12.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0002929707631371%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0002929707631371%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Escobar%20Syndrome%20Is%20a%20Prenatal%20Myasthenia%20Caused%20by%20Disruption%20of%20the%20Acetylcholine%20Receptor%20Fetal%20%5Cu03b3%20Subunit%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Katrin%22%2C%22lastName%22%3A%22Hoffmann%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Juliane%20S.%22%2C%22lastName%22%3A%22M%5Cu00fcller%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sigmar%22%2C%22lastName%22%3A%22Stricker%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Andre%22%2C%22lastName%22%3A%22Megarbane%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Anna%22%2C%22lastName%22%3A%22Rajab%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Tom%20H.%22%2C%22lastName%22%3A%22Lindner%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Monika%22%2C%22lastName%22%3A%22Cohen%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eliane%22%2C%22lastName%22%3A%22Chouery%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lynn%22%2C%22lastName%22%3A%22Adaimy%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ismat%22%2C%22lastName%22%3A%22Ghanem%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Valerie%22%2C%22lastName%22%3A%22Delague%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eugen%22%2C%22lastName%22%3A%22Boltshauser%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Beril%22%2C%22lastName%22%3A%22Talim%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rita%22%2C%22lastName%22%3A%22Horvath%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Peter%20N.%22%2C%22lastName%22%3A%22Robinson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Hanns%22%2C%22lastName%22%3A%22Lochm%5Cu00fcller%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christoph%22%2C%22lastName%22%3A%22H%5Cu00fcbner%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Stefan%22%2C%22lastName%22%3A%22Mundlos%22%7D%5D%2C%22abstractNote%22%3A%22Escobar%20syndrome%20is%20a%20form%20of%20arthrogryposis%20multiplex%20congenita%20and%20features%20joint%20contractures%2C%20pterygia%2C%20and%20respiratory%20distress.%20Similar%20findings%20occur%20in%20newborns%20exposed%20to%20nicotinergic%20acetylcholine%20receptor%20%28AChR%29%20antibodies%20from%20myasthenic%20mothers.%20We%20performed%20linkage%20studies%20in%20families%20with%20Escobar%20syndrome%20and%20identified%20eight%20mutations%20within%20the%20%5Cu03b3-subunit%20gene%20%28CHRNG%29%20of%20the%20AChR.%20Our%20functional%20studies%20show%20that%20%5Cu03b3-subunit%20mutations%20prevent%20the%20correct%20localization%20of%20the%20fetal%20AChR%20in%20human%20embryonic%20kidney%5Cu2013cell%20membranes%20and%20that%20the%20expression%20pattern%20in%20prenatal%20mice%20corresponds%20to%20the%20human%20clinical%20phenotype.%20AChRs%20have%20five%20subunits.%20Two%20%5Cu03b1%2C%20one%20%5Cu03b2%2C%20and%20one%20%5Cu03b4%20subunit%20are%20always%20present.%20By%20switching%20%5Cu03b3%20to%20%5Cu025b%20subunits%20in%20late%20fetal%20development%2C%20fetal%20AChRs%20are%20gradually%20replaced%20by%20adult%20AChRs.%20Fetal%20and%20adult%20AChRs%20are%20essential%20for%20neuromuscular%20signal%20transduction.%20In%20addition%2C%20the%20fetal%20AChRs%20seem%20to%20be%20the%20guide%20for%20the%20primary%20encounter%20of%20axon%20and%20muscle.%20Because%20of%20this%20important%20function%20in%20organogenesis%2C%20human%20mutations%20in%20the%20%5Cu03b3%20subunit%20were%20thought%20to%20be%20lethal%2C%20as%20they%20are%20in%20%5Cu03b3-knockout%20mice.%20In%20contrast%2C%20many%20mutations%20in%20other%20subunits%20have%20been%20found%20to%20be%20viable%20but%20cause%20postnatally%20persisting%20or%20beginning%20myasthenic%20syndromes.%20We%20conclude%20that%20Escobar%20syndrome%20is%20an%20inherited%20fetal%20myasthenic%20disease%20that%20also%20affects%20neuromuscular%20organogenesis.%20Because%20%5Cu03b3%20expression%20is%20restricted%20to%20early%20development%2C%20patients%20have%20no%20myasthenic%20symptoms%20later%20in%20life.%20This%20is%20the%20major%20difference%20from%20mutations%20in%20the%20other%20AChR%20subunits%20and%20the%20striking%20parallel%20to%20the%20symptoms%20found%20in%20neonates%20with%20arthrogryposis%20when%20maternal%20AChR%20auto-antibodies%20crossed%20the%20placenta%20and%20caused%20the%20transient%20inactivation%20of%20the%20AChR%20pathway.%22%2C%22date%22%3A%222006-08-01%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1086%5C%2F506257%22%2C%22ISSN%22%3A%220002-9297%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0002929707631371%22%2C%22collections%22%3A%5B%22JNM2U2A2%22%5D%2C%22dateModified%22%3A%222022-10-15T15%3A05%3A55Z%22%7D%7D%2C%7B%22key%22%3A%22EBRV45X4%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Hacohen%20et%20al.%22%2C%22parsedDate%22%3A%222015-02-01%22%2C%22numChildren%22%3A3%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BHacohen%20Y%2C%20Jacobson%20LW%2C%20Byrne%20S%2C%20Norwood%20F%2C%20Lall%20A%2C%20Robb%20S%2C%20et%20al.%20Fetal%20acetylcholine%20receptor%20inactivation%20syndrome%3A%20A%20myopathy%20due%20to%20maternal%20antibodies.%20Neurology%20-%20Neuroimmunology%20Neuroinflammation%20%5BInternet%5D.%202015%20Feb%201%20%5Bcited%202022%20Oct%2015%5D%3B2%281%29.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fnn.neurology.org%5C%2Fcontent%5C%2F2%5C%2F1%5C%2Fe57%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fnn.neurology.org%5C%2Fcontent%5C%2F2%5C%2F1%5C%2Fe57%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Fetal%20acetylcholine%20receptor%20inactivation%20syndrome%3A%20A%20myopathy%20due%20to%20maternal%20antibodies%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Yael%22%2C%22lastName%22%3A%22Hacohen%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Leslie%20W.%22%2C%22lastName%22%3A%22Jacobson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Susan%22%2C%22lastName%22%3A%22Byrne%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Fiona%22%2C%22lastName%22%3A%22Norwood%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Abhimanu%22%2C%22lastName%22%3A%22Lall%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Stephanie%22%2C%22lastName%22%3A%22Robb%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Robertino%22%2C%22lastName%22%3A%22Dilena%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Monica%22%2C%22lastName%22%3A%22Fumagalli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alfred%20Peter%22%2C%22lastName%22%3A%22Born%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Debbie%22%2C%22lastName%22%3A%22Clarke%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ming%22%2C%22lastName%22%3A%22Lim%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Angela%22%2C%22lastName%22%3A%22Vincent%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Heinz%22%2C%22lastName%22%3A%22Jungbluth%22%7D%5D%2C%22abstractNote%22%3A%22Background%3A%20Transient%20neonatal%20myasthenia%20gravis%20%28TNMG%29%20affects%20a%20proportion%20of%20infants%20born%20to%20mothers%20with%20myasthenia%20gravis%20%28MG%29.%20Symptoms%20usually%20resolve%20completely%20within%20the%20first%20few%20months%20of%20life%2C%20but%20persistent%20myopathic%20features%20have%20been%20reported%20in%20a%20few%20isolated%20cases.%5CnMethods%3A%20Here%20we%20report%208%20patients%20from%204%20families%20born%20to%20mothers%20with%20clinically%20manifest%20MG%20or%20mothers%20who%20were%20asymptomatic%20but%20had%20elevated%20acetylcholine%20receptor%20%28AChR%29%20antibody%20levels.%5CnResults%3A%20Clinical%20features%20in%20affected%20infants%20ranged%20from%20a%20mild%20predominantly%20facial%20and%20bulbar%20myopathy%20to%20arthrogryposis%20multiplex%20congenita.%20Additional%20clinical%20findings%20included%20hearing%20impairment%2C%20pyloric%20stenosis%2C%20and%20mild%20CNS%20involvement.%20In%20all%20cases%2C%20antibodies%20against%20the%20AChR%20were%20markedly%20elevated%2C%20although%20not%20always%20specific%20for%20the%20fetal%20AChR%20%5Cu03b3%20subunit.%20There%20was%20a%20correlation%20between%20maternal%20symptoms%3B%20the%20timing%2C%20intensity%2C%20and%20frequency%20of%20maternal%20treatment%3B%20and%20neonatal%20outcome.%5CnConclusions%3A%20These%20findings%20suggest%20that%20persistent%20myopathic%20features%20following%20TNMG%20may%20be%20more%20common%20than%20currently%20recognized.%20Fetal%20AChR%20inactivation%20syndrome%20should%20be%20considered%20in%20the%20differential%20diagnosis%20of%20infants%20presenting%20with%20unexplained%20myopathic%20features%2C%20in%20particular%20marked%20dysarthria%20and%20velopharyngeal%20incompetence.%20Correct%20diagnosis%20requires%20a%20high%20degree%20of%20suspicion%20if%20the%20mother%20is%20asymptomatic%20but%20is%20crucial%20considering%20the%20high%20recurrence%20risk%20for%20future%20pregnancies%20and%20the%20potentially%20treatable%20nature%20of%20this%20condition.%20Infants%20with%20a%20history%20of%20TNMG%20should%20be%20followed%20up%20for%20subtle%20myopathic%20signs%20and%20associated%20complications.%22%2C%22date%22%3A%222015%5C%2F02%5C%2F01%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1212%5C%2FNXI.0000000000000057%22%2C%22ISSN%22%3A%222332-7812%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fnn.neurology.org%5C%2Fcontent%5C%2F2%5C%2F1%5C%2Fe57%22%2C%22collections%22%3A%5B%22JNM2U2A2%22%5D%2C%22dateModified%22%3A%222022-10-15T15%3A05%3A25Z%22%7D%7D%2C%7B%22key%22%3A%22H7H64LMB%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Polyak%20et%20al.%22%2C%22parsedDate%22%3A%222015%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BPolyak%20A%2C%20Kubina%20RM%2C%20Girirajan%20S.%20Comorbidity%20of%20intellectual%20disability%20confounds%20ascertainment%20of%20autism%3A%20implications%20for%20genetic%20diagnosis.%20American%20Journal%20of%20Medical%20Genetics%20Part%20B%3A%20Neuropsychiatric%20Genetics%20%5BInternet%5D.%202015%20%5Bcited%202022%20Oct%2015%5D%3B168%287%29%3A600%26%23x2013%3B8.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1002%5C%2Fajmg.b.32338%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1002%5C%2Fajmg.b.32338%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Comorbidity%20of%20intellectual%20disability%20confounds%20ascertainment%20of%20autism%3A%20implications%20for%20genetic%20diagnosis%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Andrew%22%2C%22lastName%22%3A%22Polyak%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Richard%20M.%22%2C%22lastName%22%3A%22Kubina%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Santhosh%22%2C%22lastName%22%3A%22Girirajan%22%7D%5D%2C%22abstractNote%22%3A%22While%20recent%20studies%20suggest%20a%20converging%20role%20for%20genetic%20factors%20towards%20risk%20for%20nosologically%20distinct%20disorders%20including%20autism%2C%20intellectual%20disability%20%28ID%29%2C%20and%20epilepsy%2C%20current%20estimates%20of%20autism%20prevalence%20fail%20to%20take%20into%20account%20the%20impact%20of%20comorbidity%20of%20these%20disorders%20on%20autism%20diagnosis.%20We%20aimed%20to%20assess%20the%20effect%20of%20comorbidity%20on%20the%20diagnosis%20and%20prevalence%20of%20autism%20by%20analyzing%2011%20years%20%282000%5Cu20132010%29%20of%20special%20education%20enrollment%20data%20on%20approximately%206.2%20million%20children%20per%20year.%20We%20found%20a%20331%25%20increase%20in%20the%20prevalence%20of%20autism%20from%202000%20to%202010%20within%20special%20education%2C%20potentially%20due%20to%20a%20diagnostic%20recategorization%20from%20frequently%20comorbid%20features%20such%20as%20ID.%20The%20decrease%20in%20ID%20prevalence%20equaled%20an%20average%20of%2064.2%25%20of%20the%20increase%20of%20autism%20prevalence%20for%20children%20aged%203%5Cu201318%20years.%20The%20proportion%20of%20ID%20cases%20potentially%20undergoing%20recategorization%20to%20autism%20was%20higher%20%28P%20%3D%200.007%29%20among%20older%20children%20%2875%25%29%20than%20younger%20children%20%2848%25%29.%20Some%20US%20states%20showed%20significant%20negative%20correlations%20between%20the%20prevalence%20of%20autism%20compared%20to%20that%20of%20ID%20while%20others%20did%20not%2C%20suggesting%20state-specific%20health%20policy%20to%20be%20a%20major%20factor%20in%20categorizing%20autism.%20Further%2C%20a%20high%20frequency%20of%20autistic%20features%20was%20observed%20when%20individuals%20with%20classically%20defined%20genetic%20syndromes%20were%20evaluated%20for%20autism%20using%20standardized%20instruments.%20Our%20results%20suggest%20that%20current%20ascertainment%20practices%20are%20based%20on%20a%20single%20facet%20of%20autism-specific%20clinical%20features%20and%20do%20not%20consider%20associated%20comorbidities%20that%20may%20confound%20diagnosis.%20Longitudinal%20studies%20with%20detailed%20phenotyping%20and%20deep%20molecular%20genetic%20analyses%20are%20necessary%20to%20completely%20understand%20the%20cause%20of%20this%20complex%20disorder.%20%5Cu00a9%202015%20Wiley%20Periodicals%2C%20Inc.%22%2C%22date%22%3A%222015%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1002%5C%2Fajmg.b.32338%22%2C%22ISSN%22%3A%221552-485X%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1002%5C%2Fajmg.b.32338%22%2C%22collections%22%3A%5B%22K9BB5WBK%22%5D%2C%22dateModified%22%3A%222022-10-15T11%3A11%3A23Z%22%7D%7D%2C%7B%22key%22%3A%22WA4XIXGW%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Moore%20et%20al.%22%2C%22parsedDate%22%3A%222017-03-01%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BMoore%20CA%2C%20Staples%20JE%2C%20Dobyns%20WB%2C%20Pessoa%20A%2C%20Ventura%20CV%2C%20Fonseca%20EB%20da%2C%20et%20al.%20Characterizing%20the%20Pattern%20of%20Anomalies%20in%20Congenital%20Zika%20Syndrome%20for%20Pediatric%20Clinicians.%20JAMA%20Pediatrics%20%5BInternet%5D.%202017%20Mar%201%20%5Bcited%202022%20Oct%2015%5D%3B171%283%29%3A288%26%23x2013%3B95.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1001%5C%2Fjamapediatrics.2016.3982%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1001%5C%2Fjamapediatrics.2016.3982%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Characterizing%20the%20Pattern%20of%20Anomalies%20in%20Congenital%20Zika%20Syndrome%20for%20Pediatric%20Clinicians%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Cynthia%20A.%22%2C%22lastName%22%3A%22Moore%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22J.%20Erin%22%2C%22lastName%22%3A%22Staples%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22William%20B.%22%2C%22lastName%22%3A%22Dobyns%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Andr%5Cu00e9%22%2C%22lastName%22%3A%22Pessoa%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Camila%20V.%22%2C%22lastName%22%3A%22Ventura%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eduardo%20Borges%20da%22%2C%22lastName%22%3A%22Fonseca%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Erlane%20Marques%22%2C%22lastName%22%3A%22Ribeiro%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Liana%20O.%22%2C%22lastName%22%3A%22Ventura%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Norberto%20Nogueira%22%2C%22lastName%22%3A%22Neto%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22J.%20Fernando%22%2C%22lastName%22%3A%22Arena%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sonja%20A.%22%2C%22lastName%22%3A%22Rasmussen%22%7D%5D%2C%22abstractNote%22%3A%22Zika%20virus%20infection%20can%20be%20prenatally%20passed%20from%20a%20pregnant%20woman%20to%20her%20fetus.%20There%20is%20sufficient%20evidence%20to%20conclude%20that%20intrauterine%20Zika%20virus%20infection%20is%20a%20cause%20of%20microcephaly%20and%20serious%20brain%20anomalies%2C%20but%20the%20full%20spectrum%20of%20anomalies%20has%20not%20been%20delineated.%20To%20inform%20pediatric%20clinicians%20who%20may%20be%20called%20on%20to%20evaluate%20and%20treat%20affected%20infants%20and%20children%2C%20we%20review%20the%20most%20recent%20evidence%20to%20better%20characterize%20congenital%20Zika%20syndrome.We%20reviewed%20published%20reports%20of%20congenital%20anomalies%20occurring%20in%20fetuses%20or%20infants%20with%20presumed%20or%20laboratory-confirmed%20intrauterine%20Zika%20virus%20infection.%20We%20conducted%20a%20comprehensive%20search%20of%20the%20English%20literature%20using%20Medline%20and%20EMBASE%20for%20Zika%20from%20inception%20through%20September%2030%2C%202016.%20Congenital%20anomalies%20were%20considered%20in%20the%20context%20of%20the%20presumed%20pathogenetic%20mechanism%20related%20to%20the%20neurotropic%20properties%20of%20the%20virus.%20We%20conclude%20that%20congenital%20Zika%20syndrome%20is%20a%20recognizable%20pattern%20of%20structural%20anomalies%20and%20functional%20disabilities%20secondary%20to%20central%20and%2C%20perhaps%2C%20peripheral%20nervous%20system%20damage.%20Although%20many%20of%20the%20components%20of%20this%20syndrome%2C%20such%20as%20cognitive%2C%20sensory%2C%20and%20motor%20disabilities%2C%20are%20shared%20by%20other%20congenital%20infections%2C%20there%20are%205%20features%20that%20are%20rarely%20seen%20with%20other%20congenital%20infections%20or%20are%20unique%20to%20congenital%20Zika%20virus%20infection%3A%20%281%29%20severe%20microcephaly%20with%20partially%20collapsed%20skull%3B%20%282%29%20thin%20cerebral%20cortices%20with%20subcortical%20calcifications%3B%20%283%29%20macular%20scarring%20and%20focal%20pigmentary%20retinal%20mottling%3B%20%284%29%20congenital%20contractures%3B%20and%20%285%29%20marked%20early%20hypertonia%20and%20symptoms%20of%20extrapyramidal%20involvement.Although%20the%20full%20spectrum%20of%20adverse%20reproductive%20outcomes%20caused%20by%20Zika%20virus%20infection%20is%20not%20yet%20determined%2C%20a%20distinctive%20phenotype%5Cu2014the%20congenital%20Zika%20syndrome%5Cu2014has%20emerged.%20Recognition%20of%20this%20phenotype%20by%20clinicians%20for%20infants%20and%20children%20can%20help%20ensure%20appropriate%20etiologic%20evaluation%20and%20comprehensive%20clinical%20investigation%20to%20define%20the%20range%20of%20anomalies%20in%20an%20affected%20infant%20as%20well%20as%20determine%20essential%20follow-up%20and%20ongoing%20care.%22%2C%22date%22%3A%222017-03-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1001%5C%2Fjamapediatrics.2016.3982%22%2C%22ISSN%22%3A%222168-6203%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1001%5C%2Fjamapediatrics.2016.3982%22%2C%22collections%22%3A%5B%22YEGCBH6M%22%5D%2C%22dateModified%22%3A%222022-10-15T09%3A59%3A08Z%22%7D%7D%2C%7B%22key%22%3A%22V9JQWJ28%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Adebanjo%22%2C%22parsedDate%22%3A%222017%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BAdebanjo%20T.%20Update%3A%20Interim%20Guidance%20for%20the%20Diagnosis%2C%20Evaluation%2C%20and%20Management%20of%20Infants%20with%20Possible%20Congenital%20Zika%20Virus%20Infection%20%26%23x2014%3B%20United%20States%2C%20October%202017.%20MMWR%20Morb%20Mortal%20Wkly%20Rep%20%5BInternet%5D.%202017%20%5Bcited%202022%20Oct%2015%5D%3B66.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.cdc.gov%5C%2Fmmwr%5C%2Fvolumes%5C%2F66%5C%2Fwr%5C%2Fmm6641a1.htm%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.cdc.gov%5C%2Fmmwr%5C%2Fvolumes%5C%2F66%5C%2Fwr%5C%2Fmm6641a1.htm%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Update%3A%20Interim%20Guidance%20for%20the%20Diagnosis%2C%20Evaluation%2C%20and%20Management%20of%20Infants%20with%20Possible%20Congenital%20Zika%20Virus%20Infection%20%5Cu2014%20United%20States%2C%20October%202017%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Tolulope%22%2C%22lastName%22%3A%22Adebanjo%22%7D%5D%2C%22abstractNote%22%3A%22CDC%20has%20updated%20its%20interim%20guidance%20for%20U.S.%20health%20care%20providers%20caring%20for%20infants%20with%20possible%20congenital%20Zika%20virus%20infection%20%281%29%20in%20response%20to%20recently%20published%20updated%20guidance%20...%22%2C%22date%22%3A%222017%22%2C%22language%22%3A%22en-us%22%2C%22DOI%22%3A%2210.15585%5C%2Fmmwr.mm6641a1%22%2C%22ISSN%22%3A%220149-21951545-861X%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.cdc.gov%5C%2Fmmwr%5C%2Fvolumes%5C%2F66%5C%2Fwr%5C%2Fmm6641a1.htm%22%2C%22collections%22%3A%5B%22YEGCBH6M%22%5D%2C%22dateModified%22%3A%222022-10-15T09%3A56%3A55Z%22%7D%7D%2C%7B%22key%22%3A%22QHVIIP6J%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Koens%20et%20al.%22%2C%22parsedDate%22%3A%222018-12%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKoens%20LH%2C%20Tijssen%20MAJ%2C%20Lange%20F%2C%20Wolffenbuttel%20BHR%2C%20Rufa%20A%2C%20Zee%20DS%2C%20et%20al.%20Eye%20movement%20disorders%20and%20neurological%20symptoms%20in%20late%26%23x2010%3Bonset%20inborn%20errors%20of%20metabolism.%20Mov%20Disord%20%5BInternet%5D.%202018%20Dec%20%5Bcited%202022%20Oct%2010%5D%3B33%2812%29%3A1844%26%23x2013%3B56.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC6587951%5C%2F%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC6587951%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Eye%20movement%20disorders%20and%20neurological%20symptoms%20in%20late%5Cu2010onset%20inborn%20errors%20of%20metabolism%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lisette%20H.%22%2C%22lastName%22%3A%22Koens%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Marina%20A.J.%22%2C%22lastName%22%3A%22Tijssen%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Fiete%22%2C%22lastName%22%3A%22Lange%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Bruce%20H.R.%22%2C%22lastName%22%3A%22Wolffenbuttel%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alessandra%22%2C%22lastName%22%3A%22Rufa%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22David%20S.%22%2C%22lastName%22%3A%22Zee%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Tom%20J.%22%2C%22lastName%22%3A%22de%20Koning%22%7D%5D%2C%22abstractNote%22%3A%22Inborn%20errors%20of%20metabolism%20in%20adults%20are%20still%20largely%20unexplored.%20Despite%20the%20fact%20that%20adult%5Cu2010onset%20phenotypes%20have%20been%20known%20for%20many%20years%2C%20little%20attention%20is%20given%20to%20these%20disorders%20in%20neurological%20practice.%20The%20adult%5Cu2010onset%20presentation%20differs%20from%20childhood%5Cu2010onset%20phenotypes%2C%20often%20leading%20to%20considerable%20diagnostic%20delay.%20The%20identification%20of%20these%20patients%20at%20the%20earliest%20stage%20of%20disease%20is%20important%2C%20given%20that%20early%20treatment%20may%20prevent%20or%20lessen%20further%20brain%20damage.%20Neurological%20and%20psychiatric%20symptoms%20occur%20more%20frequently%20in%20adult%20forms.%20Abnormalities%20of%20eye%20movements%20are%20also%20common%20and%20can%20be%20the%20presenting%20sign.%20Eye%20movement%20disorders%20can%20be%20classified%20as%20central%20or%20peripheral.%20Central%20forms%20are%20frequently%20observed%20in%20lysosomal%20storage%20disorders%2C%20whereas%20peripheral%20forms%20are%20a%20key%20feature%20of%20mitochondrial%20disease.%20Furthermore%2C%20oculogyric%20crisis%20is%20an%20important%20feature%20in%20disorders%20affecting%20dopamine%20syntheses%20or%20transport.%20Ocular%20motor%20disorders%20are%20often%20not%20reported%20by%20the%20patient%2C%20and%20abnormalities%20can%20be%20easily%20overlooked%20in%20a%20general%20examination.%20In%20adults%20with%20unexplained%20psychiatric%20and%20neurological%20symptoms%2C%20a%20special%20focus%20on%20examination%20of%20eye%20movements%20can%20serve%20as%20a%20relatively%20simple%20clinical%20tool%20to%20detect%20a%20metabolic%20disorder.%20Eye%20movements%20can%20be%20easily%20quantified%20and%20analyzed%20with%20video%5Cu2010oculography%2C%20making%20them%20a%20valuable%20biomarker%20for%20following%20the%20natural%20course%20of%20disease%20or%20the%20response%20to%20therapies.%20Here%2C%20we%20review%2C%20for%20the%20first%20time%2C%20eye%20movement%20disorders%20that%20can%20occur%20in%20inborn%20errors%20of%20metabolism%2C%20with%20a%20focus%20on%20late%5Cu2010onset%20forms.%20We%20provide%20a%20step%5Cu2010by%5Cu2010step%20overview%20that%20will%20help%20clinicians%20to%20examine%20and%20interpret%20eye%20movement%20disorders.%20%5Cu00a9%202018%20The%20Authors.%20Movement%20Disorders%20published%20by%20Wiley%20Periodicals%2C%20Inc.%20on%20behalf%20of%20International%20Parkinson%20and%20Movement%20Disorder%20Society.%22%2C%22date%22%3A%222018-12%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1002%5C%2Fmds.27484%22%2C%22ISSN%22%3A%220885-3185%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC6587951%5C%2F%22%2C%22collections%22%3A%5B%22LUF9HG2X%22%5D%2C%22dateModified%22%3A%222022-10-10T14%3A48%3A14Z%22%7D%7D%2C%7B%22key%22%3A%22F4PZYNMK%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Brodsky%22%2C%22parsedDate%22%3A%222010%22%2C%22numChildren%22%3A0%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BBrodsky%20MC.%20Pediatric%20Neuro-Ophthalmology%20%5BInternet%5D.%20New%20York%2C%20NY%3A%20Springer%3B%202010%20%5Bcited%202022%20Oct%2010%5D.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2F978-0-387-69069-8%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2F978-0-387-69069-8%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22book%22%2C%22title%22%3A%22Pediatric%20Neuro-Ophthalmology%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michael%20C.%22%2C%22lastName%22%3A%22Brodsky%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222010%22%2C%22language%22%3A%22en%22%2C%22ISBN%22%3A%22978-0-387-69066-7%20978-0-387-69069-8%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2F978-0-387-69069-8%22%2C%22collections%22%3A%5B%22CEWAAZN6%22%5D%2C%22dateModified%22%3A%222022-10-10T14%3A47%3A16Z%22%7D%7D%2C%7B%22key%22%3A%22677X6MX5%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Ozonoff%20et%20al.%22%2C%22parsedDate%22%3A%222011-09%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BOzonoff%20S%2C%20Young%20GS%2C%20Carter%20A%2C%20Messinger%20D%2C%20Yirmiya%20N%2C%20Zwaigenbaum%20L%2C%20et%20al.%20Recurrence%20Risk%20for%20Autism%20Spectrum%20Disorders%3A%20A%20Baby%20Siblings%20Research%20Consortium%20Study.%20Pediatrics%20%5BInternet%5D.%202011%20Sep%20%5Bcited%202022%20Oct%2010%5D%3B128%283%29%3Ae488%26%23x2013%3B95.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC3164092%5C%2F%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC3164092%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Recurrence%20Risk%20for%20Autism%20Spectrum%20Disorders%3A%20A%20Baby%20Siblings%20Research%20Consortium%20Study%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sally%22%2C%22lastName%22%3A%22Ozonoff%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Gregory%20S.%22%2C%22lastName%22%3A%22Young%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alice%22%2C%22lastName%22%3A%22Carter%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Daniel%22%2C%22lastName%22%3A%22Messinger%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nurit%22%2C%22lastName%22%3A%22Yirmiya%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lonnie%22%2C%22lastName%22%3A%22Zwaigenbaum%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Susan%22%2C%22lastName%22%3A%22Bryson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Leslie%20J.%22%2C%22lastName%22%3A%22Carver%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22John%20N.%22%2C%22lastName%22%3A%22Constantino%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Karen%22%2C%22lastName%22%3A%22Dobkins%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ted%22%2C%22lastName%22%3A%22Hutman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jana%20M.%22%2C%22lastName%22%3A%22Iverson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rebecca%22%2C%22lastName%22%3A%22Landa%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sally%20J.%22%2C%22lastName%22%3A%22Rogers%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Marian%22%2C%22lastName%22%3A%22Sigman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Wendy%20L.%22%2C%22lastName%22%3A%22Stone%22%7D%5D%2C%22abstractNote%22%3A%22OBJECTIVE%3A%5CnThe%20recurrence%20risk%20of%20autism%20spectrum%20disorders%20%28ASD%29%20is%20estimated%20to%20be%20between%203%25%20and%2010%25%2C%20but%20previous%20research%20was%20limited%20by%20small%20sample%20sizes%20and%20biases%20related%20to%20ascertainment%2C%20reporting%2C%20and%20stoppage%20factors.%20This%20study%20used%20prospective%20methods%20to%20obtain%20an%20updated%20estimate%20of%20sibling%20recurrence%20risk%20for%20ASD.%5Cn%5CnMETHODS%3A%5CnA%20prospective%20longitudinal%20study%20of%20infants%20at%20risk%20for%20ASD%20was%20conducted%20by%20a%20multisite%20international%20network%2C%20the%20Baby%20Siblings%20Research%20Consortium.%20Infants%20%28n%20%3D%20664%29%20with%20an%20older%20biological%20sibling%20with%20ASD%20were%20followed%20from%20early%20in%20life%20to%2036%20months%2C%20when%20they%20were%20classified%20as%20having%20or%20not%20having%20ASD.%20An%20ASD%20classification%20required%20surpassing%20the%20cutoff%20of%20the%20Autism%20Diagnostic%20Observation%20Schedule%20and%20receiving%20a%20clinical%20diagnosis%20from%20an%20expert%20clinician.%5Cn%5CnRESULTS%3A%5CnA%20total%20of%2018.7%25%20of%20the%20infants%20developed%20ASD.%20Infant%20gender%20and%20the%20presence%20of%20%26gt%3B1%20older%20affected%20sibling%20were%20significant%20predictors%20of%20ASD%20outcome%2C%20and%20there%20was%20an%20almost%20threefold%20increase%20in%20risk%20for%20male%20subjects%20and%20an%20additional%20twofold%20increase%20in%20risk%20if%20there%20was%20%26gt%3B1%20older%20affected%20sibling.%20The%20age%20of%20the%20infant%20at%20study%20enrollment%2C%20the%20gender%20and%20functioning%20level%20of%20the%20infant%26%23039%3Bs%20older%20sibling%2C%20and%20other%20demographic%20factors%20did%20not%20predict%20ASD%20outcome.%5Cn%5CnCONCLUSIONS%3A%5CnThe%20sibling%20recurrence%20rate%20of%20ASD%20is%20higher%20than%20suggested%20by%20previous%20estimates.%20The%20size%20of%20the%20current%20sample%20and%20prospective%20nature%20of%20data%20collection%20minimized%20many%20limitations%20of%20previous%20studies%20of%20sibling%20recurrence.%20Clinical%20implications%2C%20including%20genetic%20counseling%2C%20are%20discussed.%22%2C%22date%22%3A%222011-9%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1542%5C%2Fpeds.2010-2825%22%2C%22ISSN%22%3A%220031-4005%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC3164092%5C%2F%22%2C%22collections%22%3A%5B%22EZBX722S%22%5D%2C%22dateModified%22%3A%222022-10-10T07%3A55%3A49Z%22%7D%7D%2C%7B%22key%22%3A%22PQ4WEZI2%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Yoo%22%2C%22parsedDate%22%3A%222022%22%2C%22numChildren%22%3A0%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BYoo%20J.%20Differential%20diagnosis%20and%20management%20of%20hyperpigmentation.%20Clinical%20and%20Experimental%20Dermatology%20%5BInternet%5D.%202022%20%5Bcited%202022%20Oct%208%5D%3B47%282%29%3A251%26%23x2013%3B8.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1111%5C%2Fced.14747%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1111%5C%2Fced.14747%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Differential%20diagnosis%20and%20management%20of%20hyperpigmentation%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22J.%22%2C%22lastName%22%3A%22Yoo%22%7D%5D%2C%22abstractNote%22%3A%22Click%20here%20for%20the%20corresponding%20questions%20to%20this%20CME%20article.%22%2C%22date%22%3A%222022%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1111%5C%2Fced.14747%22%2C%22ISSN%22%3A%221365-2230%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1111%5C%2Fced.14747%22%2C%22collections%22%3A%5B%22IRSK9CZ6%22%5D%2C%22dateModified%22%3A%222022-10-08T09%3A29%3A10Z%22%7D%7D%2C%7B%22key%22%3A%22JJVUUBUA%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Kopanos%20et%20al.%22%2C%22parsedDate%22%3A%222019-06-01%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKopanos%20C%2C%20Tsiolkas%20V%2C%20Kouris%20A%2C%20Chapple%20CE%2C%20Albarca%20Aguilera%20M%2C%20Meyer%20R%2C%20et%20al.%20VarSome%3A%20the%20human%20genomic%20variant%20search%20engine.%20Bioinformatics%20%5BInternet%5D.%202019%20Jun%201%20%5Bcited%202022%20Oct%203%5D%3B35%2811%29%3A1978%26%23x2013%3B80.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1093%5C%2Fbioinformatics%5C%2Fbty897%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1093%5C%2Fbioinformatics%5C%2Fbty897%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22VarSome%3A%20the%20human%20genomic%20variant%20search%20engine%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christos%22%2C%22lastName%22%3A%22Kopanos%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Vasilis%22%2C%22lastName%22%3A%22Tsiolkas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alexandros%22%2C%22lastName%22%3A%22Kouris%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Charles%20E%22%2C%22lastName%22%3A%22Chapple%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Monica%22%2C%22lastName%22%3A%22Albarca%20Aguilera%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Richard%22%2C%22lastName%22%3A%22Meyer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Andreas%22%2C%22lastName%22%3A%22Massouras%22%7D%5D%2C%22abstractNote%22%3A%22VarSome.com%20is%20a%20search%20engine%2C%20aggregator%20and%20impact%20analysis%20tool%20for%20human%20genetic%20variation%20and%20a%20community-driven%20project%20aiming%20at%20sharing%20global%20expertise%20on%20human%20variants.VarSome%20is%20freely%20available%20at%20http%3A%5C%2F%5C%2Fvarsome.com.Supplementary%20data%20are%20available%20at%20Bioinformatics%20online.%22%2C%22date%22%3A%222019-06-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1093%5C%2Fbioinformatics%5C%2Fbty897%22%2C%22ISSN%22%3A%221367-4803%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1093%5C%2Fbioinformatics%5C%2Fbty897%22%2C%22collections%22%3A%5B%22WN9NCB46%22%5D%2C%22dateModified%22%3A%222022-10-03T16%3A49%3A09Z%22%7D%7D%2C%7B%22key%22%3A%22NAQML864%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Tornese%20et%20al.%22%2C%22parsedDate%22%3A%222020%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BTornese%20G%2C%20Pellegrin%20MC%2C%20Barbi%20E%2C%20Ventura%20A.%20Pediatric%20endocrinology%20through%20syndromes.%20European%20Journal%20of%20Medical%20Genetics%20%5BInternet%5D.%202020%20%5Bcited%202022%20Oct%203%5D%3B63%281%29%3A103614.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Flinkinghub.elsevier.com%5C%2Fretrieve%5C%2Fpii%5C%2FS1769721218303161%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Flinkinghub.elsevier.com%5C%2Fretrieve%5C%2Fpii%5C%2FS1769721218303161%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Pediatric%20endocrinology%20through%20syndromes%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Gianluca%22%2C%22lastName%22%3A%22Tornese%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Maria%20Chiara%22%2C%22lastName%22%3A%22Pellegrin%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Egidio%22%2C%22lastName%22%3A%22Barbi%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alessandro%22%2C%22lastName%22%3A%22Ventura%22%7D%5D%2C%22abstractNote%22%3A%22In%20everyday%20practice%2C%20a%20pediatric%20endocrinologist%20will%20face%20a%20variety%20of%20different%20endocrine%20issues%20%28such%20as%20short%20or%20tall%20stature%2C%20dysthyroidism%2C%20abnormal%20pubertal%20timing%20or%20impaired%20glucose%20metabolism%29%2C%20which%20relevantly%20contribute%20to%20the%20global%20care%20of%20a%20number%20of%20syndromic%20conditions.%20On%20the%20other%20hand%2C%20the%20presence%20of%20endocrine%20features%20may%20assist%20in%20the%20diagnostic%20process%2C%20leading%20to%20final%20diagnosis%20of%20a%20syndromic%20disorder.%20The%20intention%20of%20this%20review%20is%20to%20provide%20a%20referenced%20overview%20of%20different%20genetic%20syndromes%20characterized%20by%20endocrine%20features%2C%20and%20to%20present%20a%20possible%20classification%2C%20based%20on%20whether%20the%20endocrinopathy%20or%20the%20syndrome%20is%20typically%20recognized%20first.%20Thus%2C%20the%20first%20part%20of%20the%20manuscript%20deals%20with%20the%20most%20common%20syndromes%20associated%20with%20endocrine%20dysfunctions%2C%20while%20the%20second%20part%20describes%20the%20conditions%20by%20which%20a%20syndrome%20is%20most%20frequently%20diagnosed%20after%20an%20endocrine%20finding.%20The%20aim%20is%20to%20provide%20a%20practical%20overview%20of%20the%20assessment%20of%20syndromic%20patients%2C%20so%20that%20they%20can%20be%20recognized%20and%20managed%20in%20an%20integrated%2C%20multidisciplinary%20fashion.%22%2C%22date%22%3A%2201%5C%2F2020%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1016%5C%2Fj.ejmg.2019.01.004%22%2C%22ISSN%22%3A%2217697212%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Flinkinghub.elsevier.com%5C%2Fretrieve%5C%2Fpii%5C%2FS1769721218303161%22%2C%22collections%22%3A%5B%22VXPPF34S%22%5D%2C%22dateModified%22%3A%222022-10-03T16%3A48%3A50Z%22%7D%7D%2C%7B%22key%22%3A%226I6LAZJP%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22numChildren%22%3A0%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BAnnual%20Health%20Checks%20for%20People%20with%20Intellectual%20Disabilities%20in%20General%20Practice%20%5BInternet%5D.%20%5Bcited%202022%20Oct%203%5D.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.intellectualdisability.info%5C%2Fhow-to-guides%5C%2Farticles%5C%2Fannual-health-checks-for-people-with-intellectual-disabilities-in-general-practice%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.intellectualdisability.info%5C%2Fhow-to-guides%5C%2Farticles%5C%2Fannual-health-checks-for-people-with-intellectual-disabilities-in-general-practice%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22webpage%22%2C%22title%22%3A%22Annual%20Health%20Checks%20for%20People%20with%20Intellectual%20Disabilities%20in%20General%20Practice%22%2C%22creators%22%3A%5B%5D%2C%22abstractNote%22%3A%22University%20of%20Hertfordshire%22%2C%22date%22%3A%22%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.intellectualdisability.info%5C%2Fhow-to-guides%5C%2Farticles%5C%2Fannual-health-checks-for-people-with-intellectual-disabilities-in-general-practice%22%2C%22language%22%3A%22en%22%2C%22collections%22%3A%5B%22J8JZ8I2U%22%5D%2C%22dateModified%22%3A%222022-10-03T16%3A46%3A28Z%22%7D%7D%2C%7B%22key%22%3A%22X2S3FZEA%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Lagorio%20et%20al.%22%2C%22parsedDate%22%3A%222022-08-01%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BLagorio%20I%2C%20Brunelli%20L%2C%20Striano%20P.%20Paroxysmal%20Nonepileptic%20Events%20in%20Children%3A%20A%20Video%20Gallery%20and%20a%20Guide%20for%20Differential%20Diagnosis.%20Neurology%3A%20Clinical%20Practice%20%5BInternet%5D.%202022%20Aug%201%20%5Bcited%202022%20Oct%203%5D%3B12%284%29%3A320%26%23x2013%3B7.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fcp.neurology.org%5C%2Fcontent%5C%2F12%5C%2F4%5C%2F320%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fcp.neurology.org%5C%2Fcontent%5C%2F12%5C%2F4%5C%2F320%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Paroxysmal%20Nonepileptic%20Events%20in%20Children%3A%20A%20Video%20Gallery%20and%20a%20Guide%20for%20Differential%20Diagnosis%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ilaria%22%2C%22lastName%22%3A%22Lagorio%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lorenzo%22%2C%22lastName%22%3A%22Brunelli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pasquale%22%2C%22lastName%22%3A%22Striano%22%7D%5D%2C%22abstractNote%22%3A%22Purpose%20of%20Review%20Paroxysmal%20nonepileptic%20events%20%28PNEEs%29%20are%20a%20heterogenous%20group%20of%20time-limited%20events%2C%20characterized%20by%20changes%20in%20motor%20or%20behavioral%20activity%20beginning%20abruptly%20and%20ending%20in%20a%20short%20time.%20Owing%20to%20their%20manifestation%2C%20these%20conditions%20can%20clinically%20simulate%20seizures.%5CnRecent%20Findings%20These%20episodes%20belong%20to%20different%20categories%2C%20including%20syncopal%20events%2C%20psychiatric%20disorders%2C%20and%20movement%20disorders.%20PNEEs%20are%20a%20common%20cause%20of%20diagnostic%20mistakes%20and%20families%26%23039%3B%20concerns%2C%20and%20the%20risk%20of%20useless%20and%20sometimes%20even%20injurious%20treatment%20is%20considerable.%20The%20high%20frequency%20of%20these%20manifestations%20in%20clinical%20practice%20makes%20PNEEs%20a%20diagnostic%20challenge%20for%20clinicians.%5CnSummary%20This%20review%20is%20focused%20on%20the%20distinctive%20clinical%20findings%20and%20treatment%20of%20PNEEs.%20Illustrative%20video%20recordings%20of%20the%20PNEEs%20and%20a%20video%20collection%20as%20a%20support%20tool%20for%20differential%20diagnosis%20are%20provided.%22%2C%22date%22%3A%222022%5C%2F08%5C%2F01%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1212%5C%2FCPJ.0000000000001171%22%2C%22ISSN%22%3A%222163-0402%2C%202163-0933%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fcp.neurology.org%5C%2Fcontent%5C%2F12%5C%2F4%5C%2F320%22%2C%22collections%22%3A%5B%22BFFSCGQP%22%5D%2C%22dateModified%22%3A%222022-10-03T16%3A43%3A29Z%22%7D%7D%2C%7B%22key%22%3A%22YLQY2H3N%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22de%20Freitas%20et%20al.%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3Bde%20Freitas%20FD%2C%20Pimenta%20S%2C%20Soares%20S%2C%20Gonzaga%20D%2C%20Vaz-Matos%20I%2C%20Prior%20C.%20El%20papel%20de%20los%20cannabinoides%20en%20los%20trastornos%20del%20neurodesarrollo%20de%20ni%26%23xF1%3Bos%20y%20adolescentes.%20Rev%20Neurol.%20%3A9.%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22El%20papel%20de%20los%20cannabinoides%20en%20los%20trastornos%20del%20neurodesarrollo%20de%20ni%5Cu00f1os%20y%20adolescentes%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Francisca%20Dias%22%2C%22lastName%22%3A%22de%20Freitas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sofia%22%2C%22lastName%22%3A%22Pimenta%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sara%22%2C%22lastName%22%3A%22Soares%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Diana%22%2C%22lastName%22%3A%22Gonzaga%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22In%5Cu00eas%22%2C%22lastName%22%3A%22Vaz-Matos%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Catarina%22%2C%22lastName%22%3A%22Prior%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%22%22%2C%22language%22%3A%22es%22%2C%22DOI%22%3A%22%22%2C%22ISSN%22%3A%22%22%2C%22url%22%3A%22%22%2C%22collections%22%3A%5B%22HWIG92V4%22%5D%2C%22dateModified%22%3A%222022-10-03T16%3A42%3A37Z%22%7D%7D%2C%7B%22key%22%3A%22FTGW32SA%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Marwaha%20et%20al.%22%2C%22parsedDate%22%3A%222022-02-28%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BMarwaha%20S%2C%20Knowles%20JW%2C%20Ashley%20EA.%20A%20guide%20for%20the%20diagnosis%20of%20rare%20and%20undiagnosed%20disease%3A%20beyond%20the%20exome.%20Genome%20Medicine%20%5BInternet%5D.%202022%20Feb%2028%20%5Bcited%202022%20Oct%203%5D%3B14%281%29%3A23.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1186%5C%2Fs13073-022-01026-w%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1186%5C%2Fs13073-022-01026-w%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22A%20guide%20for%20the%20diagnosis%20of%20rare%20and%20undiagnosed%20disease%3A%20beyond%20the%20exome%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Shruti%22%2C%22lastName%22%3A%22Marwaha%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Joshua%20W.%22%2C%22lastName%22%3A%22Knowles%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Euan%20A.%22%2C%22lastName%22%3A%22Ashley%22%7D%5D%2C%22abstractNote%22%3A%22Rare%20diseases%20affect%2030%20million%20people%20in%20the%20USA%20and%20more%20than%20300%5Cu2013400%20million%20worldwide%2C%20often%20causing%20chronic%20illness%2C%20disability%2C%20and%20premature%20death.%20Traditional%20diagnostic%20techniques%20rely%20heavily%20on%20heuristic%20approaches%2C%20coupling%20clinical%20experience%20from%20prior%20rare%20disease%20presentations%20with%20the%20medical%20literature.%20A%20large%20number%20of%20rare%20disease%20patients%20remain%20undiagnosed%20for%20years%20and%20many%20even%20die%20without%20an%20accurate%20diagnosis.%20In%20recent%20years%2C%20gene%20panels%2C%20microarrays%2C%20and%20exome%20sequencing%20have%20helped%20to%20identify%20the%20molecular%20cause%20of%20such%20rare%20and%20undiagnosed%20diseases.%20These%20technologies%20have%20allowed%20diagnoses%20for%20a%20sizable%20proportion%20%2825%5Cu201335%25%29%20of%20undiagnosed%20patients%2C%20often%20with%20actionable%20findings.%20However%2C%20a%20large%20proportion%20of%20these%20patients%20remain%20undiagnosed.%20In%20this%20review%2C%20we%20focus%20on%20technologies%20that%20can%20be%20adopted%20if%20exome%20sequencing%20is%20unrevealing.%20We%20discuss%20the%20benefits%20of%20sequencing%20the%20whole%20genome%20and%20the%20additional%20benefit%20that%20may%20be%20offered%20by%20long-read%20technology%2C%20pan-genome%20reference%2C%20transcriptomics%2C%20metabolomics%2C%20proteomics%2C%20and%20methyl%20profiling.%20We%20highlight%20computational%20methods%20to%20help%20identify%20regionally%20distant%20patients%20with%20similar%20phenotypes%20or%20similar%20genetic%20mutations.%20Finally%2C%20we%20describe%20approaches%20to%20automate%20and%20accelerate%20genomic%20analysis.%20The%20strategies%20discussed%20here%20are%20intended%20to%20serve%20as%20a%20guide%20for%20clinicians%20and%20researchers%20in%20the%20next%20steps%20when%20encountering%20patients%20with%20non-diagnostic%20exomes.%22%2C%22date%22%3A%222022-02-28%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1186%5C%2Fs13073-022-01026-w%22%2C%22ISSN%22%3A%221756-994X%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1186%5C%2Fs13073-022-01026-w%22%2C%22collections%22%3A%5B%22N2ARV9VB%22%5D%2C%22dateModified%22%3A%222022-10-03T16%3A37%3A52Z%22%7D%7D%2C%7B%22key%22%3A%22MENBMLMF%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Patel%20et%20al.%22%2C%22parsedDate%22%3A%222020-12-29%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BPatel%20RA%2C%20Hall%20DA%2C%20Eichenseer%20S%2C%20Bailey%20M.%20Movement%20Disorders%20and%20Hematologic%20Diseases.%20Mov%20Disord%20Clin%20Pract%20%5BInternet%5D.%202020%20Dec%2029%20%5Bcited%202022%20Oct%203%5D%3B8%282%29%3A193%26%23x2013%3B207.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC7853188%5C%2F%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC7853188%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Movement%20Disorders%20and%20Hematologic%20Diseases%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Roshni%20Abee%22%2C%22lastName%22%3A%22Patel%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Deborah%20A.%22%2C%22lastName%22%3A%22Hall%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sheila%22%2C%22lastName%22%3A%22Eichenseer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Meagan%22%2C%22lastName%22%3A%22Bailey%22%7D%5D%2C%22abstractNote%22%3A%22Background%5CnMovement%20disorders%20can%20be%20associated%20with%20or%20caused%20by%20hematological%20abnormalities.%20The%20objective%20of%20this%20review%20is%20to%20highlight%20features%20that%20will%20aid%20in%20the%20clinician%26%23039%3Bs%20recognition%20and%20treatment%20of%20these%20disorders.%5Cn%5CnMethods%5CnMESH%20terms%20relevant%20to%20movement%20disorders%20and%20hematologic%20diseases%20were%20searched%20to%20identify%20conditions%20included%20in%20this%20narrative%2C%20educational%20review.%5Cn%5CnResults%5CnSeveral%20conditions%20were%20identified%2C%20and%20they%20were%20organized%20by%20hematologic%20categories%20to%20include%20red%20blood%20cell%20abnormalities%2C%20white%20blood%20cell%20abnormalities%2C%20disorders%20of%20clotting%20and%20bleeding%2C%20hematologic%20malignancies%2C%20and%20others.%5Cn%5CnConclusions%5CnThis%20review%20will%20increase%20providers%5Cu2019%20understanding%20of%20disorders%20that%20include%20movement%20disorders%20and%20hematologic%20abnormalities.%20Basic%20hematologic%20laboratories%20can%20aid%20in%20assessment%20of%20these%20disorders%2C%20to%20include%20complete%20blood%20count%5C%2Fhemogram%20and%20peripheral%20blood%20smear.%20Recognition%20is%20key%2C%20especially%20in%20the%20setting%20of%20underlying%20malignancy%2C%20vitamin%20deficiency%2C%20or%20other%20disorder%20in%20which%20treatment%20is%20available.%22%2C%22date%22%3A%222020-12-29%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1002%5C%2Fmdc3.13129%22%2C%22ISSN%22%3A%222330-1619%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC7853188%5C%2F%22%2C%22collections%22%3A%5B%22FRRGKHP5%22%5D%2C%22dateModified%22%3A%222022-10-03T16%3A21%3A56Z%22%7D%7D%2C%7B%22key%22%3A%224XQ7J44S%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Girirajan%20et%20al.%22%2C%22parsedDate%22%3A%222012-10-04%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BGirirajan%20S%2C%20Rosenfeld%20JA%2C%20Coe%20BP%2C%20Parikh%20S%2C%20Friedman%20N%2C%20Goldstein%20A%2C%20et%20al.%20Phenotypic%20Heterogeneity%20of%20Genomic%20Disorders%20and%20Rare%20Copy-Number%20Variants.%20New%20England%20Journal%20of%20Medicine%20%5BInternet%5D.%202012%20Oct%204%20%5Bcited%202022%20Oct%203%5D%3B367%2814%29%3A1321%26%23x2013%3B31.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1056%5C%2FNEJMoa1200395%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1056%5C%2FNEJMoa1200395%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Phenotypic%20Heterogeneity%20of%20Genomic%20Disorders%20and%20Rare%20Copy-Number%20Variants%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Santhosh%22%2C%22lastName%22%3A%22Girirajan%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jill%20A.%22%2C%22lastName%22%3A%22Rosenfeld%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Bradley%20P.%22%2C%22lastName%22%3A%22Coe%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sumit%22%2C%22lastName%22%3A%22Parikh%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Neil%22%2C%22lastName%22%3A%22Friedman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Amy%22%2C%22lastName%22%3A%22Goldstein%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Robyn%20A.%22%2C%22lastName%22%3A%22Filipink%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Juliann%20S.%22%2C%22lastName%22%3A%22McConnell%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Brad%22%2C%22lastName%22%3A%22Angle%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Wendy%20S.%22%2C%22lastName%22%3A%22Meschino%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Marjan%20M.%22%2C%22lastName%22%3A%22Nezarati%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alexander%22%2C%22lastName%22%3A%22Asamoah%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Kelly%20E.%22%2C%22lastName%22%3A%22Jackson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Gordon%20C.%22%2C%22lastName%22%3A%22Gowans%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Judith%20A.%22%2C%22lastName%22%3A%22Martin%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Erin%20P.%22%2C%22lastName%22%3A%22Carmany%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22David%20W.%22%2C%22lastName%22%3A%22Stockton%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rhonda%20E.%22%2C%22lastName%22%3A%22Schnur%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lynette%20S.%22%2C%22lastName%22%3A%22Penney%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Donna%20M.%22%2C%22lastName%22%3A%22Martin%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Salmo%22%2C%22lastName%22%3A%22Raskin%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Kathleen%22%2C%22lastName%22%3A%22Leppig%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Heidi%22%2C%22lastName%22%3A%22Thiese%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rosemarie%22%2C%22lastName%22%3A%22Smith%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Erika%22%2C%22lastName%22%3A%22Aberg%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Dmitriy%20M.%22%2C%22lastName%22%3A%22Niyazov%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Luis%20F.%22%2C%22lastName%22%3A%22Escobar%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Dima%22%2C%22lastName%22%3A%22El-Khechen%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Kisha%20D.%22%2C%22lastName%22%3A%22Johnson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Robert%20R.%22%2C%22lastName%22%3A%22Lebel%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Kiana%22%2C%22lastName%22%3A%22Siefkas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Susie%22%2C%22lastName%22%3A%22Ball%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Natasha%22%2C%22lastName%22%3A%22Shur%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Marianne%22%2C%22lastName%22%3A%22McGuire%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Campbell%20K.%22%2C%22lastName%22%3A%22Brasington%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22J.%20Edward%22%2C%22lastName%22%3A%22Spence%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Laura%20S.%22%2C%22lastName%22%3A%22Martin%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Carol%22%2C%22lastName%22%3A%22Clericuzio%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Blake%20C.%22%2C%22lastName%22%3A%22Ballif%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lisa%20G.%22%2C%22lastName%22%3A%22Shaffer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Evan%20E.%22%2C%22lastName%22%3A%22Eichler%22%7D%5D%2C%22abstractNote%22%3A%22Genomic%20rearrangements%20are%20an%20important%20source%20of%20genetic%20and%20phenotypic%20variation.%20Rare%2C%20recurrent%20copy-number%20variants%20of%20pathogenic%20significance%2C%20termed%20genomic%20disorders%2C%20were%20originally%20identified%20in%20persons%20with%20a%20characteristic%20set%20of%20clinically%20recognizable%20features%2C%20such%20as%20the%20Smith%5Cu2013Magenis%20syndrome%2C%20the%20Sotos%20syndrome%2C%20and%20the%20Williams%5Cu2013Beuren%20syndrome.%20Although%20unexplained%20phenotypic%20variation%20and%20differences%20in%20severity%20have%20long%20been%20recognized%20among%20patients%20with%20the%20same%20genomic%20disorder%2C1%5Cu20135%20comparatively%20recent%20discoveries%20of%20potentially%20pathogenic%20copy-number%20variants%20have%20broadened%20the%20phenotypic%20range%20associated%20with%20a%20given%20variant%20to%20include%20entirely%20distinct%20diseases.%20High-throughput%20analyses%20of%20patient%20populations%20have%20implicated%20the%20same%20copy-number%20variants%20.%20.%20.%22%2C%22date%22%3A%222012-10-04%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1056%5C%2FNEJMoa1200395%22%2C%22ISSN%22%3A%220028-4793%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1056%5C%2FNEJMoa1200395%22%2C%22collections%22%3A%5B%22X4RCMSQI%22%5D%2C%22dateModified%22%3A%222022-10-03T08%3A10%3A14Z%22%7D%7D%2C%7B%22key%22%3A%22AT3R5PNY%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Bharath%20et%20al.%22%2C%22parsedDate%22%3A%222010%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BBharath%20R%2C%20Unnikrishnan%20AG%2C%20Thampy%20MV%2C%20Anilkumar%20A%2C%20Nisha%20B%2C%20Praveen%20VP%2C%20et%20al.%20Turner%20syndrome%20and%20its%20variants.%20Indian%20J%20Pediatr%20%5BInternet%5D.%202010%20%5Bcited%202022%20Sep%2024%5D%3B77%282%29%3A193%26%23x2013%3B5.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2Fs12098-009-0226-7%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2Fs12098-009-0226-7%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Turner%20syndrome%20and%20its%20variants%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22R.%22%2C%22lastName%22%3A%22Bharath%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22A.%20G.%22%2C%22lastName%22%3A%22Unnikrishnan%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22M.%20V.%22%2C%22lastName%22%3A%22Thampy%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alka%22%2C%22lastName%22%3A%22Anilkumar%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22B.%22%2C%22lastName%22%3A%22Nisha%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22V.%20P.%22%2C%22lastName%22%3A%22Praveen%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Vasantha%22%2C%22lastName%22%3A%22Nair%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22R.%20V.%22%2C%22lastName%22%3A%22Jayakumar%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Harish%22%2C%22lastName%22%3A%22Kumar%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222%5C%2F2010%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1007%5C%2Fs12098-009-0226-7%22%2C%22ISSN%22%3A%220019-5456%2C%200973-7693%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2Fs12098-009-0226-7%22%2C%22collections%22%3A%5B%22AMP8AUIM%22%5D%2C%22dateModified%22%3A%222022-09-24T12%3A49%3A58Z%22%7D%7D%2C%7B%22key%22%3A%228MVW57D9%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Liehr%22%2C%22parsedDate%22%3A%222016-01-22%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BLiehr%20T.%20Cytogenetically%20visible%20copy%20number%20variations%20%28CG-CNVs%29%20in%20banding%20and%20molecular%20cytogenetics%20of%20human%3B%20about%20heteromorphisms%20and%20euchromatic%20variants.%20Mol%20Cytogenet%20%5BInternet%5D.%202016%20Jan%2022%20%5Bcited%202022%20Sep%2024%5D%3B9%3A5.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC4724132%5C%2F%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC4724132%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Cytogenetically%20visible%20copy%20number%20variations%20%28CG-CNVs%29%20in%20banding%20and%20molecular%20cytogenetics%20of%20human%3B%20about%20heteromorphisms%20and%20euchromatic%20variants%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Thomas%22%2C%22lastName%22%3A%22Liehr%22%7D%5D%2C%22abstractNote%22%3A%22Background%5CnCopy%20number%20variations%20%28CNVs%29%20having%20no%20%28obvious%29%20clinical%20effects%20were%20rediscovered%20as%20major%20part%20of%20human%20genome%20in%202004.%20However%2C%20for%20every%20cytogeneticist%20microscopically%20visible%20harmless%20CNVs%20%28CG-CNVs%29%20are%20well%20known%20since%20decades.%20Harmless%20CG-CNVs%20can%20be%20present%20as%20heterochromatic%20or%20even%20as%20euchromatic%20variants%20in%20clinically%20healthy%20persons.%5Cn%5CnResults%5CnHere%20I%20provide%20a%20review%20on%20what%20is%20known%20today%20on%20the%20still%20too%20little%20studied%20harmless%20human%20CG-CNVs%2C%20point%20out%20which%20can%20be%20mixed%20up%20with%20clinically%20relevant%20pathological%20CG-CNVs%20and%20shortly%20discuss%20that%20the%20artificial%20separation%20of%20euchromatic%20submicroscopic%20CNVs%20%28MG-CNVs%29%20and%20euchromatic%20CG-CNVs%20is%20no%20longer%20timely.%5Cn%5CnConclusion%5CnOverall%2C%20neither%20so-called%20harmless%20heterochromatic%20nor%20so-called%20harmless%20euchromatic%20CG-CNVs%20are%20considered%20enough%20in%20evaluation%20of%20routine%20cytogenetic%20analysis%20and%20reporting.%20This%20holds%20especially%20true%20when%20bearing%20in%20mind%20the%20so-called%20two-hit%20model%20suggesting%20that%20combination%20of%20per%20se%20harmless%20CNVs%20may%20lead%20to%20clinical%20aberrations%20if%20they%20are%20present%20together%20in%20one%20patient.%22%2C%22date%22%3A%222016-1-22%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1186%5C%2Fs13039-016-0216-1%22%2C%22ISSN%22%3A%221755-8166%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC4724132%5C%2F%22%2C%22collections%22%3A%5B%22AMP8AUIM%22%5D%2C%22dateModified%22%3A%222022-09-24T11%3A00%3A22Z%22%7D%7D%2C%7B%22key%22%3A%226SL9DY7W%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BNothing%26%23x2019%3Bs%20for%20sure%2C%20that%26%23x2019%3Bs%20for%20sure%3A%20Evaluating%20variants%20of%20uncertain%20significance%20%7C%20Beyond%20the%20Ion%20Channel%20%5BInternet%5D.%20%5Bcited%202022%20Sep%2022%5D.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fepilepsygenetics.net%5C%2F2016%5C%2F08%5C%2F11%5C%2Fnothings-for-sure-thats-for-sure-evaluating-variants-of-uncertain-significance%5C%2F%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fepilepsygenetics.net%5C%2F2016%5C%2F08%5C%2F11%5C%2Fnothings-for-sure-thats-for-sure-evaluating-variants-of-uncertain-significance%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22blogPost%22%2C%22title%22%3A%22Nothing%5Cu2019s%20for%20sure%2C%20that%5Cu2019s%20for%20sure%3A%20Evaluating%20variants%20of%20uncertain%20significance%20%7C%20Beyond%20the%20Ion%20Channel%22%2C%22creators%22%3A%5B%5D%2C%22abstractNote%22%3A%22%22%2C%22blogTitle%22%3A%22%22%2C%22date%22%3A%22%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fepilepsygenetics.net%5C%2F2016%5C%2F08%5C%2F11%5C%2Fnothings-for-sure-thats-for-sure-evaluating-variants-of-uncertain-significance%5C%2F%22%2C%22language%22%3A%22en-US%22%2C%22collections%22%3A%5B%22N2ARV9VB%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A29%3A43Z%22%7D%7D%2C%7B%22key%22%3A%22RI6TWRHS%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Vaz-Drago%20et%20al.%22%2C%22parsedDate%22%3A%222017%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BVaz-Drago%20R%2C%20Cust%26%23xF3%3Bdio%20N%2C%20Carmo-Fonseca%20M.%20Deep%20intronic%20mutations%20and%20human%20disease.%20Hum%20Genet%20%5BInternet%5D.%202017%20%5Bcited%202020%20May%203%5D%3B136%289%29%3A1093%26%23x2013%3B111.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2Fs00439-017-1809-4%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2Fs00439-017-1809-4%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Deep%20intronic%20mutations%20and%20human%20disease%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rita%22%2C%22lastName%22%3A%22Vaz-Drago%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22No%5Cu00e9lia%22%2C%22lastName%22%3A%22Cust%5Cu00f3dio%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Maria%22%2C%22lastName%22%3A%22Carmo-Fonseca%22%7D%5D%2C%22abstractNote%22%3A%22Next-generation%20sequencing%20has%20revolutionized%20clinical%20diagnostic%20testing.%20Yet%2C%20for%20a%20substantial%20proportion%20of%20patients%2C%20sequence%20information%20restricted%20to%20exons%20and%20exon%5Cu2013intron%20boundaries%20fails%20to%20identify%20the%20genetic%20cause%20of%20the%20disease.%20Here%20we%20review%20evidence%20from%20mRNA%20analysis%20and%20entire%20genomic%20sequencing%20indicating%20that%20pathogenic%20mutations%20can%20occur%20deep%20within%20the%20introns%20of%20over%2075%20disease-associated%20genes.%20Deleterious%20DNA%20variants%20located%20more%20than%20100%20base%20pairs%20away%20from%20exon%5Cu2013intron%20junctions%20most%20commonly%20lead%20to%20pseudo-exon%20inclusion%20due%20to%20activation%20of%20non-canonical%20splice%20sites%20or%20changes%20in%20splicing%20regulatory%20elements.%20Additionally%2C%20deep%20intronic%20mutations%20can%20disrupt%20transcription%20regulatory%20motifs%20and%20non-coding%20RNA%20genes.%20This%20review%20aims%20to%20highlight%20the%20importance%20of%20studying%20variation%20in%20deep%20intronic%20sequence%20as%20a%20cause%20of%20monogenic%20disorders%20as%20well%20as%20hereditary%20cancer%20syndromes.%22%2C%22date%22%3A%229%5C%2F2017%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1007%5C%2Fs00439-017-1809-4%22%2C%22ISSN%22%3A%220340-6717%2C%201432-1203%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Flink.springer.com%5C%2F10.1007%5C%2Fs00439-017-1809-4%22%2C%22collections%22%3A%5B%227XAESF88%22%2C%22B8DDX5EW%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A27%3A25Z%22%7D%7D%2C%7B%22key%22%3A%22MBL4PJ2Q%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Barros%20et%20al.%22%2C%22parsedDate%22%3A%222018%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BBarros%20FS%2C%20Marussi%20VHR%2C%20Amaral%20LLF%2C%20da%20Rocha%20AJ%2C%20Campos%20CMS%2C%20Freitas%20LF%2C%20et%20al.%20The%20Rare%20Neurocutaneous%20Disorders%3A%20Update%20on%20Clinical%2C%20Molecular%2C%20and%20Neuroimaging%20Features.%20Topics%20in%20Magnetic%20Resonance%20Imaging%20%5BInternet%5D.%202018%20%5Bcited%202019%20Apr%2028%5D%3B27%286%29%3A433%26%23x2013%3B62.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2FInsights.ovid.com%5C%2Fcrossref%3Fan%3D00002142-201812000-00004%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2FInsights.ovid.com%5C%2Fcrossref%3Fan%3D00002142-201812000-00004%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22The%20Rare%20Neurocutaneous%20Disorders%3A%20Update%20on%20Clinical%2C%20Molecular%2C%20and%20Neuroimaging%20Features%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Felipe%20S.%22%2C%22lastName%22%3A%22Barros%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Victor%20Hugo%20R.%22%2C%22lastName%22%3A%22Marussi%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22L%5Cu00e1zaro%20L.F.%22%2C%22lastName%22%3A%22Amaral%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ant%5Cu00f4nio%20Jos%5Cu00e9%22%2C%22lastName%22%3A%22da%20Rocha%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christiane%20M.S.%22%2C%22lastName%22%3A%22Campos%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Leonardo%20F.%22%2C%22lastName%22%3A%22Freitas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Thierry%20A.G.M.%22%2C%22lastName%22%3A%22Huisman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Bruno%20P.%22%2C%22lastName%22%3A%22Soares%22%7D%5D%2C%22abstractNote%22%3A%22Phakomatoses%2C%20also%20known%20as%20neurocutaneous%20disorders%2C%20comprise%20a%20vast%20number%20of%20entities%20that%20predominantly%20affect%20structures%20originated%20from%20the%20ectoderm%20such%20as%20the%20central%20nervous%20system%20and%20the%20skin%2C%20but%20also%20the%20mesoderm%2C%20particularly%20the%20vascular%20system.%20Extensive%20literature%20exists%20about%20the%20most%20common%20phakomatoses%2C%20namely%20neurofibromatosis%2C%20tuberous%20sclerosis%2C%20von%20Hippel-Lindau%20and%20Sturge-Weber%20syndrome.%20However%2C%20recent%20developments%20in%20the%20understanding%20of%20the%20molecular%20underpinnings%20of%20less%20common%20phakomatoses%20have%20sparked%20interest%20in%20these%20disorders.%20In%20this%20article%2C%20we%20review%20the%20clinical%20features%2C%20current%20pathogenesis%2C%20and%20modern%20neuroimaging%20findings%20of%20melanophakomatoses%2C%20vascular%20phakomatoses%2C%20and%20other%20rare%20neurocutaneous%20syndromes%20that%20may%20also%20include%20tissue%20overgrowth%20or%20neoplastic%20predisposition.%22%2C%22date%22%3A%2212%5C%2F2018%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1097%5C%2FRMR.0000000000000185%22%2C%22ISSN%22%3A%220899-3459%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2FInsights.ovid.com%5C%2Fcrossref%3Fan%3D00002142-201812000-00004%22%2C%22collections%22%3A%5B%223E3R62NP%22%2C%22KPCKVNIG%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A27%3A23Z%22%7D%7D%2C%7B%22key%22%3A%22JYSMUMP3%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Gabriele%20et%20al.%22%2C%22parsedDate%22%3A%222018-06-08%22%2C%22numChildren%22%3A3%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BGabriele%20M%2C%20Lopez%20Tobon%20A%2C%20D%26%23x2019%3BAgostino%20G%2C%20Testa%20G.%20The%20chromatin%20basis%20of%20neurodevelopmental%20disorders%3A%20Rethinking%20dysfunction%20along%20the%20molecular%20and%20temporal%20axes.%20Progress%20in%20Neuro-Psychopharmacology%20and%20Biological%20Psychiatry%20%5BInternet%5D.%202018%20Jun%208%20%5Bcited%202019%20Oct%205%5D%3B84%3A306%26%23x2013%3B27.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0278584617305389%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0278584617305389%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22The%20chromatin%20basis%20of%20neurodevelopmental%20disorders%3A%20Rethinking%20dysfunction%20along%20the%20molecular%20and%20temporal%20axes%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michele%22%2C%22lastName%22%3A%22Gabriele%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alejandro%22%2C%22lastName%22%3A%22Lopez%20Tobon%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Giuseppe%22%2C%22lastName%22%3A%22D%27Agostino%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Giuseppe%22%2C%22lastName%22%3A%22Testa%22%7D%5D%2C%22abstractNote%22%3A%22The%20complexity%20of%20the%20human%20brain%20emerges%20from%20a%20long%20and%20finely%20tuned%20developmental%20process%20orchestrated%20by%20the%20crosstalk%20between%20genome%20and%20environment.%20Vis%20%5Cu00e0%20vis%20other%20species%2C%20the%20human%20brain%20displays%20unique%20functional%20and%20morphological%20features%20that%20result%20from%20this%20extensive%20developmental%20process%20that%20is%2C%20unsurprisingly%2C%20highly%20vulnerable%20to%20both%20genetically%20and%20environmentally%20induced%20alterations.%20One%20of%20the%20most%20striking%20outcomes%20of%20the%20recent%20surge%20of%20sequencing-based%20studies%20on%20neurodevelopmental%20disorders%20%28NDDs%29%20is%20the%20emergence%20of%20chromatin%20regulation%20as%20one%20of%20the%20two%20domains%20most%20affected%20by%20causative%20mutations%20or%20Copy%20Number%20Variations%20besides%20synaptic%20function%2C%20whose%20involvement%20had%20been%20largely%20predicted%20for%20obvious%20reasons.%20These%20observations%20place%20chromatin%20dysfunction%20at%20the%20top%20of%20the%20molecular%20pathways%20hierarchy%20that%20ushers%20in%20a%20sizeable%20proportion%20of%20NDDs%20and%20that%20manifest%20themselves%20through%20synaptic%20dysfunction%20and%20recurrent%20systemic%20clinical%20manifestation.%20Here%20we%20undertake%20a%20conceptual%20investigation%20of%20chromatin%20dysfunction%20in%20NDDs%20with%20the%20aim%20of%20systematizing%20the%20available%20evidence%20in%20a%20new%20framework%3A%20first%2C%20we%20tease%20out%20the%20developmental%20vulnerabilities%20in%20human%20corticogenesis%20as%20a%20structuring%20entry%20point%20into%20the%20causation%20of%20NDDs%3B%20second%2C%20we%20provide%20a%20much%20needed%20clarification%20of%20the%20multiple%20meanings%20and%20explanatory%20frameworks%20revolving%20around%20%26quot%3Bepigenetics%26quot%3B%2C%20highlighting%20those%20that%20are%20most%20relevant%20for%20the%20analysis%20of%20these%20disorders%3B%20finally%20we%20go%20in-depth%20into%20paradigmatic%20examples%20of%20NDD-causing%20chromatin%20dysregulation%2C%20with%20a%20special%20focus%20on%20human%20experimental%20models%20and%20datasets.%22%2C%22date%22%3A%22June%208%2C%202018%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1016%5C%2Fj.pnpbp.2017.12.013%22%2C%22ISSN%22%3A%220278-5846%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS0278584617305389%22%2C%22collections%22%3A%5B%22VB9PLJM9%22%2C%227ZJWGN9I%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A27%3A13Z%22%7D%7D%2C%7B%22key%22%3A%226Y64ZDU5%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Guerra%20and%20Cacabelos%22%2C%22parsedDate%22%3A%222019%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BGuerra%20J%2C%20Cacabelos%20R.%20Genomics%20of%20speech%20and%20language%20disorders.%20JTGG%20%5BInternet%5D.%202019%20%5Bcited%202020%20Sep%2025%5D%3B%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fjtggjournal.com%5C%2Farticle%5C%2Fview%5C%2F3118%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fjtggjournal.com%5C%2Farticle%5C%2Fview%5C%2F3118%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Genomics%20of%20speech%20and%20language%20disorders%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Joaquin%22%2C%22lastName%22%3A%22Guerra%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ramon%22%2C%22lastName%22%3A%22Cacabelos%22%7D%5D%2C%22abstractNote%22%3A%22Multiple%20factors%20involve%20speech%20and%20language.%20Investigating%20animal%20models%2C%20mainly%20through%20songbirds%2C%20has%20allowed%20a%20better%20understanding%20of%20the%20verbal%20communication%20process.%20Speech%20disorders%2C%20such%20as%20childhood%20apraxia%20of%20speech%2C%20dysarthria%20or%20stuttering%2C%20along%20with%20language%20disorders%2C%20like%20aphasia%2C%20dyslexia%20or%20developmental%20language%20disorder%20are%20the%20main%20examples.%20More%20complex%20syndromes%20such%20as%20Autism-spectrum%20disorders%2C%20Down%5Cu2019s%20syndrome%20or%20Fragile%20X%20syndrome%20have%20more%20variable%20features.%20Genetic%20factors%2C%20such%20as%20hereditary%20or%20de%20novo%20mutations%20may%20influence%20the%20development%20of%20all%20of%20these%20conditions.%20Besides%2C%20most%20of%20speech%20and%20language%20disorders%20are%20implicated%20in%20neurodevelopment%20with%20molecular%20mechanisms%20and%20pathways%20that%20interact%20with%20each%20other%2C%20and%20there%20may%20be%20co-morbidity%20with%20other%20communication%20disorders%20or%20phenotypes%20unrelated%20to%20communication.%20Genes%20with%20heterogeneous%20functions%20in%20speech%20and%20language%20such%20as%20FOXP1%20%2C%20FOXP2%20%2C%20KIAA0319%20%2C%20ROBO1%20%2C%20APOE%20or%20CNTNAP2%20are%20some%20examples.%20Epigenetic%20factors%2C%20especially%20microRNAs%2C%20influence%20the%20expressiveness.%20The%20genomics%20of%20these%20disorders%20allows%20us%20to%20understand%20language%20acquisition%2C%20carry%20out%20early%20detection%20strategies%2C%20genetic%20counseling%20and%20optimize%20future%20treatments%2C%20not%20only%20in%20communication%20disorders%20but%20also%20the%20neurological%20alterations%20that%20incorporate%20these%20mutations.%22%2C%22date%22%3A%222019%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.20517%5C%2Fjtgg.2018.03%22%2C%22ISSN%22%3A%22%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fjtggjournal.com%5C%2Farticle%5C%2Fview%5C%2F3118%22%2C%22collections%22%3A%5B%2293W8FE6J%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A27%3A06Z%22%7D%7D%2C%7B%22key%22%3A%22ZPCHWLAP%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Truty%20et%20al.%22%2C%22parsedDate%22%3A%222019%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BTruty%20R%2C%20Paul%20J%2C%20Kennemer%20M%2C%20Lincoln%20SE%2C%20Olivares%20E%2C%20Nussbaum%20RL%2C%20et%20al.%20Prevalence%20and%20properties%20of%20intragenic%20copy-number%20variation%20in%20Mendelian%20disease%20genes.%20Genet%20Med%20%5BInternet%5D.%202019%20%5Bcited%202019%20Sep%203%5D%3B21%281%29%3A114%26%23x2013%3B23.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.nature.com%5C%2Farticles%5C%2Fs41436-018-0033-5%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.nature.com%5C%2Farticles%5C%2Fs41436-018-0033-5%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Prevalence%20and%20properties%20of%20intragenic%20copy-number%20variation%20in%20Mendelian%20disease%20genes%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rebecca%22%2C%22lastName%22%3A%22Truty%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Joshua%22%2C%22lastName%22%3A%22Paul%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michael%22%2C%22lastName%22%3A%22Kennemer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Stephen%20E.%22%2C%22lastName%22%3A%22Lincoln%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eric%22%2C%22lastName%22%3A%22Olivares%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Robert%20L.%22%2C%22lastName%22%3A%22Nussbaum%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Swaroop%22%2C%22lastName%22%3A%22Aradhya%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%221%5C%2F2019%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1038%5C%2Fs41436-018-0033-5%22%2C%22ISSN%22%3A%221098-3600%2C%201530-0366%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.nature.com%5C%2Farticles%5C%2Fs41436-018-0033-5%22%2C%22collections%22%3A%5B%227XAESF88%22%2C%22X543ERCQ%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A27%3A02Z%22%7D%7D%2C%7B%22key%22%3A%223YIHHINZ%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Garone%20et%20al.%22%2C%22parsedDate%22%3A%222020-05-20%22%2C%22numChildren%22%3A4%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BGarone%20G%2C%20Capuano%20A%2C%20Travaglini%20L%2C%20Graziola%20F%2C%20Stregapede%20F%2C%20Zanni%20G%2C%20et%20al.%20Clinical%20and%20Genetic%20Overview%20of%20Paroxysmal%20Movement%20Disorders%20and%20Episodic%20Ataxias.%20IJMS%20%5BInternet%5D.%202020%20May%2020%20%5Bcited%202021%20Sep%202%5D%3B21%2810%29%3A3603.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F1422-0067%5C%2F21%5C%2F10%5C%2F3603%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F1422-0067%5C%2F21%5C%2F10%5C%2F3603%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Clinical%20and%20Genetic%20Overview%20of%20Paroxysmal%20Movement%20Disorders%20and%20Episodic%20Ataxias%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Giacomo%22%2C%22lastName%22%3A%22Garone%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alessandro%22%2C%22lastName%22%3A%22Capuano%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lorena%22%2C%22lastName%22%3A%22Travaglini%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Federica%22%2C%22lastName%22%3A%22Graziola%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Fabrizia%22%2C%22lastName%22%3A%22Stregapede%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ginevra%22%2C%22lastName%22%3A%22Zanni%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Federico%22%2C%22lastName%22%3A%22Vigevano%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Enrico%22%2C%22lastName%22%3A%22Bertini%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Francesco%22%2C%22lastName%22%3A%22Nicita%22%7D%5D%2C%22abstractNote%22%3A%22Paroxysmal%20movement%20disorders%20%28PMDs%29%20are%20rare%20neurological%20diseases%20typically%20manifesting%20with%20intermittent%20attacks%20of%20abnormal%20involuntary%20movements.%20Two%20main%20categories%20of%20PMDs%20are%20recognized%20based%20on%20the%20phenomenology%3A%20Paroxysmal%20dyskinesias%20%28PxDs%29%20are%20characterized%20by%20transient%20episodes%20hyperkinetic%20movement%20disorders%2C%20while%20attacks%20of%20cerebellar%20dysfunction%20are%20the%20hallmark%20of%20episodic%20ataxias%20%28EAs%29.%20From%20an%20etiological%20point%20of%20view%2C%20both%20primary%20%28genetic%29%20and%20secondary%20%28acquired%29%20causes%20of%20PMDs%20are%20known.%20Recognition%20and%20diagnosis%20of%20PMDs%20is%20based%20on%20personal%20and%20familial%20medical%20history%2C%20physical%20examination%2C%20detailed%20reconstruction%20of%20ictal%20phenomenology%2C%20neuroimaging%2C%20and%20genetic%20analysis.%20Neurophysiological%20or%20laboratory%20tests%20are%20reserved%20for%20selected%20cases.%20Genetic%20knowledge%20of%20PMDs%20has%20been%20largely%20incremented%20by%20the%20advent%20of%20next%20generation%20sequencing%20%28NGS%29%20methodologies.%20The%20wide%20number%20of%20genes%20involved%20in%20the%20pathogenesis%20of%20PMDs%20re%5Cufb02ects%20a%20high%20complexity%20of%20molecular%20bases%20of%20neurotransmission%20in%20cerebellar%20and%20basal%20ganglia%20circuits.%20In%20consideration%20of%20the%20broad%20genetic%20and%20phenotypic%20heterogeneity%2C%20a%20NGS%20approach%20by%20targeted%20panel%20for%20movement%20disorders%2C%20clinical%20or%20whole%20exome%20sequencing%20should%20be%20preferred%2C%20whenever%20possible%2C%20to%20a%20single%20gene%20approach%2C%20in%20order%20to%20increase%20diagnostic%20rate.%20This%20review%20is%20focused%20on%20clinical%20and%20genetic%20features%20of%20PMDs%20with%20the%20aim%20to%20%281%29%20help%20clinicians%20to%20recognize%2C%20diagnose%20and%20treat%20patients%20with%20PMDs%20as%20well%20as%20to%20%282%29%20provide%20an%20overview%20of%20genes%20and%20molecular%20mechanisms%20underlying%20these%20intriguing%20neurogenetic%20disorders.%22%2C%22date%22%3A%222020-05-20%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.3390%5C%2Fijms21103603%22%2C%22ISSN%22%3A%221422-0067%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F1422-0067%5C%2F21%5C%2F10%5C%2F3603%22%2C%22collections%22%3A%5B%22PG6XL6QT%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A26%3A54Z%22%7D%7D%2C%7B%22key%22%3A%224N7CVVVJ%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Karbassi%20et%20al.%22%2C%22parsedDate%22%3A%222016%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKarbassi%20I%2C%20Maston%20GA%2C%20Love%20A%2C%20DiVincenzo%20C%2C%20Braastad%20CD%2C%20Elzinga%20CD%2C%20et%20al.%20A%20Standardized%20DNA%20Variant%20Scoring%20System%20for%20Pathogenicity%20Assessments%20in%20Mendelian%20Disorders.%20Human%20Mutation%20%5BInternet%5D.%202016%20%5Bcited%202022%20Sep%2022%5D%3B37%281%29%3A127%26%23x2013%3B34.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1002%5C%2Fhumu.22918%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1002%5C%2Fhumu.22918%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22A%20Standardized%20DNA%20Variant%20Scoring%20System%20for%20Pathogenicity%20Assessments%20in%20Mendelian%20Disorders%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Izabela%22%2C%22lastName%22%3A%22Karbassi%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Glenn%20A.%22%2C%22lastName%22%3A%22Maston%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Angela%22%2C%22lastName%22%3A%22Love%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christina%22%2C%22lastName%22%3A%22DiVincenzo%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Corey%20D.%22%2C%22lastName%22%3A%22Braastad%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christopher%20D.%22%2C%22lastName%22%3A%22Elzinga%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alison%20R.%22%2C%22lastName%22%3A%22Bright%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Domenic%22%2C%22lastName%22%3A%22Previte%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ke%22%2C%22lastName%22%3A%22Zhang%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Charles%20M.%22%2C%22lastName%22%3A%22Rowland%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michele%22%2C%22lastName%22%3A%22McCarthy%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jennifer%20L.%22%2C%22lastName%22%3A%22Lapierre%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Felicita%22%2C%22lastName%22%3A%22Dubois%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Katelyn%20A.%22%2C%22lastName%22%3A%22Medeiros%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sat%20Dev%22%2C%22lastName%22%3A%22Batish%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jeffrey%22%2C%22lastName%22%3A%22Jones%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Khalida%22%2C%22lastName%22%3A%22Liaquat%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Carol%20A.%22%2C%22lastName%22%3A%22Hoffman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Malgorzata%22%2C%22lastName%22%3A%22Jaremko%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Zhenyuan%22%2C%22lastName%22%3A%22Wang%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Weimin%22%2C%22lastName%22%3A%22Sun%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Arlene%22%2C%22lastName%22%3A%22Buller-Burckle%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Charles%20M.%22%2C%22lastName%22%3A%22Strom%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Steven%20B.%22%2C%22lastName%22%3A%22Keiles%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Joseph%20J.%22%2C%22lastName%22%3A%22Higgins%22%7D%5D%2C%22abstractNote%22%3A%22We%20developed%20a%20rules-based%20scoring%20system%20to%20classify%20DNA%20variants%20into%20five%20categories%20including%20pathogenic%2C%20likely%20pathogenic%2C%20variant%20of%20uncertain%20significance%20%28VUS%29%2C%20likely%20benign%2C%20and%20benign.%20Over%2016%2C500%20pathogenicity%20assessments%20on%2011%2C894%20variants%20from%20338%20genes%20were%20analyzed%20for%20pathogenicity%20based%20on%20prediction%20tools%2C%20population%20frequency%2C%20co-occurrence%2C%20segregation%2C%20and%20functional%20studies%20collected%20from%20internal%20and%20external%20sources.%20Scores%20were%20calculated%20by%20trained%20scientists%20using%20a%20quantitative%20framework%20that%20assigned%20differential%20weighting%20to%20these%20five%20types%20of%20data.%20We%20performed%20descriptive%20and%20comparative%20statistics%20on%20the%20dataset%20and%20tested%20interobserver%20concordance%20among%20the%20trained%20scientists.%20Private%20variants%20defined%20as%20variants%20found%20within%20single%20families%20%28n%20%3D%205%2C182%29%2C%20were%20either%20VUS%20%2880.5%25%3B%20n%20%3D%204%2C169%29%20or%20likely%20pathogenic%20%2819.5%25%3B%20n%20%3D%201%2C013%29.%20The%20remaining%20variants%20%28n%20%3D%206%2C712%29%20were%20VUS%20%2838.4%25%3B%20n%20%3D%202%2C577%29%20or%20likely%20benign%5C%2Fbenign%20%2834.7%25%3B%20n%20%3D%202%2C327%29%20or%20likely%20pathogenic%5C%2Fpathogenic%20%2826.9%25%2C%20n%20%3D%201%2C808%29.%20Exact%20agreement%20between%20the%20trained%20scientists%20on%20the%20final%20variant%20score%20was%2098.5%25%20%5B95%25%20confidence%20interval%20%28CI%29%20%2898.0%2C%2098.9%29%5D%20with%20an%20interobserver%20consistency%20of%2097%25%20%5B95%25%20CI%20%2891.5%2C%2099.4%29%5D.%20Variant%20scores%20were%20stable%20and%20showed%20increasing%20odds%20of%20being%20in%20agreement%20with%20new%20data%20when%20re-evaluated%20periodically.%20This%20carefully%20curated%2C%20standardized%20variant%20pathogenicity%20scoring%20system%20provides%20reliable%20pathogenicity%20scores%20for%20DNA%20variants%20encountered%20in%20a%20clinical%20laboratory%20setting.%22%2C%22date%22%3A%222016%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1002%5C%2Fhumu.22918%22%2C%22ISSN%22%3A%221098-1004%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fonlinelibrary.wiley.com%5C%2Fdoi%5C%2Fabs%5C%2F10.1002%5C%2Fhumu.22918%22%2C%22collections%22%3A%5B%227XAESF88%22%5D%2C%22dateModified%22%3A%222022-09-22T17%3A26%3A27Z%22%7D%7D%2C%7B%22key%22%3A%22EQ9J2U6P%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Beetz%20and%20Bauer%22%2C%22parsedDate%22%3A%222020%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BBeetz%20C%2C%20Bauer%20P.%20Dual%20genetic%20diagnoses%20-%20underappreciated%20%26%23x201C%3Bdouble%20trouble.%26%23x201D%3B%20JBCGenetics%20%5BInternet%5D.%202020%20%5Bcited%202022%20Sep%2013%5D%3B52%26%23x2013%3B3.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ejmanager.com%5C%2Ffulltextpdf.php%3Fmno%3D135141%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ejmanager.com%5C%2Ffulltextpdf.php%3Fmno%3D135141%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Dual%20genetic%20diagnoses%20-%20underappreciated%20%5C%22double%20trouble%5C%22%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christian%22%2C%22lastName%22%3A%22Beetz%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Peter%22%2C%22lastName%22%3A%22Bauer%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222020%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.24911%5C%2FJBCGenetics%5C%2F183-1600154983%22%2C%22ISSN%22%3A%221658-807X%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ejmanager.com%5C%2Ffulltextpdf.php%3Fmno%3D135141%22%2C%22collections%22%3A%5B%22N2ARV9VB%22%5D%2C%22dateModified%22%3A%222022-09-13T18%3A59%3A25Z%22%7D%7D%2C%7B%22key%22%3A%22XV3J9PPL%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Posey%20et%20al.%22%2C%22parsedDate%22%3A%222017-01-05%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BPosey%20JE%2C%20Harel%20T%2C%20Liu%20P%2C%20Rosenfeld%20JA%2C%20James%20RA%2C%20Coban%20Akdemir%20ZH%2C%20et%20al.%20Resolution%20of%20Disease%20Phenotypes%20Resulting%20from%20Multilocus%20Genomic%20Variation.%20New%20England%20Journal%20of%20Medicine%20%5BInternet%5D.%202017%20Jan%205%20%5Bcited%202022%20Sep%2013%5D%3B376%281%29%3A21%26%23x2013%3B31.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1056%5C%2FNEJMoa1516767%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1056%5C%2FNEJMoa1516767%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Resolution%20of%20Disease%20Phenotypes%20Resulting%20from%20Multilocus%20Genomic%20Variation%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jennifer%20E.%22%2C%22lastName%22%3A%22Posey%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Tamar%22%2C%22lastName%22%3A%22Harel%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pengfei%22%2C%22lastName%22%3A%22Liu%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jill%20A.%22%2C%22lastName%22%3A%22Rosenfeld%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Regis%20A.%22%2C%22lastName%22%3A%22James%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Zeynep%20H.%22%2C%22lastName%22%3A%22Coban%20Akdemir%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Magdalena%22%2C%22lastName%22%3A%22Walkiewicz%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Weimin%22%2C%22lastName%22%3A%22Bi%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rui%22%2C%22lastName%22%3A%22Xiao%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Yan%22%2C%22lastName%22%3A%22Ding%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Fan%22%2C%22lastName%22%3A%22Xia%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Arthur%20L.%22%2C%22lastName%22%3A%22Beaudet%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Donna%20M.%22%2C%22lastName%22%3A%22Muzny%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Richard%20A.%22%2C%22lastName%22%3A%22Gibbs%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eric%22%2C%22lastName%22%3A%22Boerwinkle%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Christine%20M.%22%2C%22lastName%22%3A%22Eng%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22V.%20Reid%22%2C%22lastName%22%3A%22Sutton%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Chad%20A.%22%2C%22lastName%22%3A%22Shaw%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sharon%20E.%22%2C%22lastName%22%3A%22Plon%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Yaping%22%2C%22lastName%22%3A%22Yang%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22James%20R.%22%2C%22lastName%22%3A%22Lupski%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222017-01-05%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1056%5C%2FNEJMoa1516767%22%2C%22ISSN%22%3A%220028-4793%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1056%5C%2FNEJMoa1516767%22%2C%22collections%22%3A%5B%22N2ARV9VB%22%5D%2C%22dateModified%22%3A%222022-09-13T18%3A30%3A19Z%22%7D%7D%2C%7B%22key%22%3A%22AM8XGDGQ%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Koutroumanidis%20et%20al.%22%2C%22parsedDate%22%3A%222017-12-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKoutroumanidis%20M%2C%20Arzimanoglou%20A%2C%20Caraballo%20R%2C%20Goyal%20S%2C%20Kaminska%20A%2C%20Laoprasert%20P%2C%20et%20al.%20The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%202%29.%20Epileptic%20Disorders%20%5BInternet%5D.%202017%20Dec%201%20%5Bcited%202022%20Sep%2012%5D%3B19%284%29%3A385%26%23x2013%3B437.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218%5C%2Farticle.phtml%3Ftab%3Dtexte%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218%5C%2Farticle.phtml%3Ftab%3Dtexte%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%202%29%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michalis%22%2C%22lastName%22%3A%22Koutroumanidis%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alexis%22%2C%22lastName%22%3A%22Arzimanoglou%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Roberto%22%2C%22lastName%22%3A%22Caraballo%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sushma%22%2C%22lastName%22%3A%22Goyal%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Anna%22%2C%22lastName%22%3A%22Kaminska%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pramote%22%2C%22lastName%22%3A%22Laoprasert%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Hirokazu%22%2C%22lastName%22%3A%22Oguni%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Guido%22%2C%22lastName%22%3A%22Rubboli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22William%22%2C%22lastName%22%3A%22Tatum%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pierre%22%2C%22lastName%22%3A%22Thomas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eugen%22%2C%22lastName%22%3A%22Trinka%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Luca%22%2C%22lastName%22%3A%22Vignatelli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Solomon%20L.%22%2C%22lastName%22%3A%22Mosh%5Cu00e9%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222017-12-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1684%5C%2Fepd.2017.0952%22%2C%22ISSN%22%3A%221294-9361%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218%5C%2Farticle.phtml%3Ftab%3Dtexte%22%2C%22collections%22%3A%5B%22IVUN2VXI%22%5D%2C%22dateModified%22%3A%222022-09-12T15%3A25%3A11Z%22%7D%7D%2C%7B%22key%22%3A%22DKTQN32G%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Koutroumanidis%20et%20al.%22%2C%22parsedDate%22%3A%222017-09-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKoutroumanidis%20M%2C%20Arzimanoglou%20A%2C%20Caraballo%20R%2C%20Goyal%20S%2C%20Kaminska%20A%2C%20Laoprasert%20P%2C%20et%20al.%20The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%201%29.%20Epileptic%20Disorders%20%5BInternet%5D.%202017%20Sep%201%20%5Bcited%202022%20Sep%2012%5D%3B19%283%29%3A233%26%23x2013%3B98.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508%5C%2Farticle.phtml%3Ftab%3Dtexte%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508%5C%2Farticle.phtml%3Ftab%3Dtexte%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%201%29%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michalis%22%2C%22lastName%22%3A%22Koutroumanidis%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alexis%22%2C%22lastName%22%3A%22Arzimanoglou%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Roberto%22%2C%22lastName%22%3A%22Caraballo%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sushma%22%2C%22lastName%22%3A%22Goyal%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Anna%22%2C%22lastName%22%3A%22Kaminska%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pramote%22%2C%22lastName%22%3A%22Laoprasert%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Hirokazu%22%2C%22lastName%22%3A%22Oguni%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Guido%22%2C%22lastName%22%3A%22Rubboli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22William%22%2C%22lastName%22%3A%22Tatum%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pierre%22%2C%22lastName%22%3A%22Thomas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eugen%22%2C%22lastName%22%3A%22Trinka%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Luca%22%2C%22lastName%22%3A%22Vignatelli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Solomon%20L.%22%2C%22lastName%22%3A%22Mosh%5Cu00e9%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222017-09-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1684%5C%2Fepd.2017.0935%22%2C%22ISSN%22%3A%221294-9361%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508%5C%2Farticle.phtml%3Ftab%3Dtexte%22%2C%22collections%22%3A%5B%22IVUN2VXI%22%5D%2C%22dateModified%22%3A%222022-09-12T15%3A25%3A01Z%22%7D%7D%2C%7B%22key%22%3A%228YPTTNYP%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Bronkhorst%20et%20al.%22%2C%22parsedDate%22%3A%222022-09-03%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BBronkhorst%20AJ%2C%20Ungerer%20V%2C%20Oberhofer%20A%2C%20Gabriel%20S%2C%20Polatoglou%20E%2C%20Randeu%20H%2C%20et%20al.%20New%20Perspectives%20on%20the%20Importance%20of%20Cell-Free%20DNA%20Biology.%20Diagnostics%20%5BInternet%5D.%202022%20Sep%203%20%5Bcited%202022%20Sep%2012%5D%3B12%289%29%3A2147.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F2075-4418%5C%2F12%5C%2F9%5C%2F2147%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F2075-4418%5C%2F12%5C%2F9%5C%2F2147%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22New%20Perspectives%20on%20the%20Importance%20of%20Cell-Free%20DNA%20Biology%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Abel%20J.%22%2C%22lastName%22%3A%22Bronkhorst%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Vida%22%2C%22lastName%22%3A%22Ungerer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Angela%22%2C%22lastName%22%3A%22Oberhofer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sophie%22%2C%22lastName%22%3A%22Gabriel%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eleni%22%2C%22lastName%22%3A%22Polatoglou%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Hannah%22%2C%22lastName%22%3A%22Randeu%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Carsten%22%2C%22lastName%22%3A%22Uhlig%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Heiko%22%2C%22lastName%22%3A%22Pfister%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Zsuzsanna%22%2C%22lastName%22%3A%22Mayer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Stefan%22%2C%22lastName%22%3A%22Holdenrieder%22%7D%5D%2C%22abstractNote%22%3A%22Body%20%5Cufb02uids%20are%20constantly%20replenished%20with%20a%20population%20of%20genetically%20diverse%20cell-free%20DNA%20%28cfDNA%29%20fragments%2C%20representing%20a%20vast%20reservoir%20of%20information%20re%5Cufb02ecting%20real-time%20changes%20in%20the%20host%20and%20metagenome.%20As%20many%20body%20%5Cufb02uids%20can%20be%20collected%20non-invasively%20in%20a%20one-off%20and%20serial%20fashion%2C%20this%20reservoir%20can%20be%20tapped%20to%20develop%20assays%20for%20the%20diagnosis%2C%20prognosis%2C%20and%20monitoring%20of%20wide-ranging%20pathologies%2C%20such%20as%20solid%20tumors%2C%20fetal%20genetic%20abnormalities%2C%20rejected%20organ%20transplants%2C%20infections%2C%20and%20potentially%20many%20others.%20The%20translation%20of%20cfDNA%20research%20into%20useful%20clinical%20tests%20is%20gaining%20momentum%2C%20with%20recent%20progress%20being%20driven%20by%20rapidly%20evolving%20preanalytical%20and%20analytical%20procedures%2C%20integrated%20bioinformatics%2C%20and%20machine%20learning%20algorithms.%20Yet%2C%20despite%20these%20spectacular%20advances%2C%20cfDNA%20remains%20a%20very%20challenging%20analyte%20due%20to%20its%20immense%20heterogeneity%20and%20%5Cufb02uctuation%20in%20vivo.%20It%20is%20increasingly%20recognized%20that%20high-%5Cufb01delity%20reconstruction%20of%20the%20information%20stored%20in%20cfDNA%2C%20and%20in%20turn%20the%20development%20of%20tests%20that%20are%20%5Cufb01t%20for%20clinical%20roll-out%2C%20requires%20a%20much%20deeper%20understanding%20of%20both%20the%20physico-chemical%20features%20of%20cfDNA%20and%20the%20biological%2C%20physiological%2C%20lifestyle%2C%20and%20environmental%20factors%20that%20modulate%20it.%20This%20is%20a%20daunting%20task%2C%20but%20with%20signi%5Cufb01cant%20upsides.%20In%20this%20review%20we%20showed%20how%20expanded%20knowledge%20on%20cfDNA%20biology%20and%20faithful%20reverse-engineering%20of%20cfDNA%20samples%20promises%20to%20%28i%29%20augment%20the%20sensitivity%20and%20speci%5Cufb01city%20of%20existing%20cfDNA%20assays%3B%20%28ii%29%20expand%20the%20repertoire%20of%20disease-speci%5Cufb01c%20cfDNA%20markers%2C%20thereby%20leading%20to%20the%20development%20of%20increasingly%20powerful%20assays%3B%20%28iii%29%20reshape%20personal%20molecular%20medicine%3B%20and%20%28iv%29%20have%20an%20unprecedented%20impact%20on%20genetics%20research.%22%2C%22date%22%3A%222022-09-03%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.3390%5C%2Fdiagnostics12092147%22%2C%22ISSN%22%3A%222075-4418%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F2075-4418%5C%2F12%5C%2F9%5C%2F2147%22%2C%22collections%22%3A%5B%22IN6395D8%22%5D%2C%22dateModified%22%3A%222022-09-12T15%3A24%3A04Z%22%7D%7D%2C%7B%22key%22%3A%22U2YLTKQB%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Friedman%22%2C%22parsedDate%22%3A%222022-04-21%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BFriedman%20JM.%20Neurofibromatosis%201%20%5BInternet%5D.%20University%20of%20Washington%2C%20Seattle%3B%202022%20%5Bcited%202022%20Sep%2012%5D.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fbooks%5C%2FNBK1109%5C%2F%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fbooks%5C%2FNBK1109%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22book%22%2C%22title%22%3A%22Neurofibromatosis%201%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jan%20M.%22%2C%22lastName%22%3A%22Friedman%22%7D%5D%2C%22abstractNote%22%3A%22Neurofibromatosis%201%20%28NF1%29%20is%20a%20multisystem%20disorder%20characterized%20by%20multiple%20caf%5Cu00e9%20au%20lait%20macules%2C%20intertriginous%20freckling%2C%20multiple%20cutaneous%20neurofibromas%2C%20and%20learning%20disability%20or%20behavior%20problems.%20About%20half%20of%20people%20with%20NF1%20have%20plexiform%20neurofibromas%2C%20but%20most%20are%20internal%20and%20not%20suspected%20clinically.%20Plexiform%20neurofibromas%20can%20cause%20pain%2C%20neurologic%20deficits%2C%20and%20abnormalities%20of%20involved%20or%20adjacent%20structures.%20Less%20common%20but%20potentially%20more%20serious%20manifestations%20include%20optic%20nerve%20and%20other%20central%20nervous%20system%20gliomas%2C%20malignant%20peripheral%20nerve%20sheath%20tumors%2C%20scoliosis%2C%20tibial%20dysplasia%2C%20vasculopathy%2C%20and%20gastrointestinal%2C%20endocrine%2C%20or%20pulmonary%20disease.%22%2C%22date%22%3A%222022%5C%2F04%5C%2F21%22%2C%22language%22%3A%22en%22%2C%22ISBN%22%3A%22%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fbooks%5C%2FNBK1109%5C%2F%22%2C%22collections%22%3A%5B%229PE95M6Q%22%5D%2C%22dateModified%22%3A%222022-09-12T14%3A16%3A51Z%22%7D%7D%2C%7B%22key%22%3A%223QPEXIWK%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BMyths%20of%20Human%20Genetics%3A%20Introduction%20%5BInternet%5D.%20%5Bcited%202022%20Sep%206%5D.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fudel.edu%5C%2F~mcdonald%5C%2Fmythintro.html%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fudel.edu%5C%2F~mcdonald%5C%2Fmythintro.html%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22webpage%22%2C%22title%22%3A%22Myths%20of%20Human%20Genetics%3A%20Introduction%22%2C%22creators%22%3A%5B%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%22%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fudel.edu%5C%2F~mcdonald%5C%2Fmythintro.html%22%2C%22language%22%3A%22%22%2C%22collections%22%3A%5B%22WN9NCB46%22%5D%2C%22dateModified%22%3A%222022-09-06T18%3A11%3A00Z%22%7D%7D%2C%7B%22key%22%3A%22GB3XKZN6%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Zafar%20et%20al.%22%2C%22parsedDate%22%3A%222020-11-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BZafar%20S%2C%20Doria%20J%2C%20Karceski%20S.%20Should%20we%20standardize%20the%20EEG%20classification%20of%20mild%2C%20moderate%2C%20and%20severe%20cerebral%20dysfunction%3F%20Epilepsy%20Behav%20%5BInternet%5D.%202020%20Nov%201%20%5Bcited%202022%20Sep%206%5D%3B112.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.epilepsybehavior.com%5C%2Farticle%5C%2FS1525-5050%252820%252930511-4%5C%2Ffulltext%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.epilepsybehavior.com%5C%2Farticle%5C%2FS1525-5050%252820%252930511-4%5C%2Ffulltext%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Should%20we%20standardize%20the%20EEG%20classification%20of%20mild%2C%20moderate%2C%20and%20severe%20cerebral%20dysfunction%3F%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Saman%22%2C%22lastName%22%3A%22Zafar%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Joseph%22%2C%22lastName%22%3A%22Doria%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Steven%22%2C%22lastName%22%3A%22Karceski%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222020-11-01%22%2C%22language%22%3A%22English%22%2C%22DOI%22%3A%2210.1016%5C%2Fj.yebeh.2020.107332%22%2C%22ISSN%22%3A%221525-5050%2C%201525-5069%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.epilepsybehavior.com%5C%2Farticle%5C%2FS1525-5050%252820%252930511-4%5C%2Ffulltext%22%2C%22collections%22%3A%5B%22GVAPV5VA%22%5D%2C%22dateModified%22%3A%222022-09-06T07%3A39%3A28Z%22%7D%7D%2C%7B%22key%22%3A%22PADBQ3HE%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Marks%20et%20al.%22%2C%22parsedDate%22%3A%222020-08-07%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BMarks%20K%2C%20Coutinho%20E%2C%20Vincent%20A.%20Maternal-Autoantibody-Related%20%28MAR%29%20Autism%3A%20Identifying%20Neuronal%20Antigens%20and%20Approaching%20Prospects%20for%20Intervention.%20JCM%20%5BInternet%5D.%202020%20Aug%207%20%5Bcited%202022%20Aug%2031%5D%3B9%288%29%3A2564.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F2077-0383%5C%2F9%5C%2F8%5C%2F2564%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F2077-0383%5C%2F9%5C%2F8%5C%2F2564%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Maternal-Autoantibody-Related%20%28MAR%29%20Autism%3A%20Identifying%20Neuronal%20Antigens%20and%20Approaching%20Prospects%20for%20Intervention%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Katya%22%2C%22lastName%22%3A%22Marks%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ester%22%2C%22lastName%22%3A%22Coutinho%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Angela%22%2C%22lastName%22%3A%22Vincent%22%7D%5D%2C%22abstractNote%22%3A%22Recent%20studies%20indicate%20the%20existence%20of%20a%20maternal-autoantibody-related%20subtype%20of%20autism%20spectrum%20disorder%20%28ASD%29.%20To%20date%2C%20a%20large%20number%20of%20studies%20have%20focused%20on%20describing%20patterns%20of%20brain-reactive%20serum%20antibodies%20in%20maternal-autoantibody-related%20%28MAR%29%20autism%20and%20some%20have%20described%20attempts%20to%20de%5Cufb01ne%20the%20antigenic%20targets.%20This%20article%20describes%20evidence%20on%20MAR%20autism%20and%20the%20various%20autoantibodies%20that%20have%20been%20implicated.%20Among%20other%20possibilities%2C%20antibodies%20to%20neuronal%20surface%20protein%20Contactin%20Associated%20Protein%202%20%28CASPR2%29%20have%20been%20found%20more%20frequently%20in%20mothers%20of%20children%20with%20neurodevelopmental%20disorders%20or%20autism%2C%20and%20two%20independent%20experimental%20studies%20have%20shown%20pathogenicity%20in%20mice.%20The%20N-methyl-D-aspartate%20receptor%20%28NMDAR%29%20is%20another%20possible%20target%20for%20maternal%20antibodies%20as%20demonstrated%20in%20mice.%20Here%2C%20we%20discuss%20the%20growing%20evidence%2C%20discuss%20issues%20regarding%20biomarker%20de%5Cufb01nition%2C%20and%20summarise%20the%20therapeutic%20approaches%20that%20might%20be%20used%20to%20reduce%20or%20prevent%20the%20transfer%20of%20pathogenic%20maternal%20antibodies.%22%2C%22date%22%3A%222020-08-07%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.3390%5C%2Fjcm9082564%22%2C%22ISSN%22%3A%222077-0383%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.mdpi.com%5C%2F2077-0383%5C%2F9%5C%2F8%5C%2F2564%22%2C%22collections%22%3A%5B%22R5L5SLLE%22%5D%2C%22dateModified%22%3A%222022-08-31T10%3A44%3A01Z%22%7D%7D%2C%7B%22key%22%3A%226HUZD7E3%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Watkins%20et%20al.%22%2C%22parsedDate%22%3A%222011-11-23%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BWatkins%20CE%2C%20Litchfield%20J%2C%20Song%20E%2C%20Jaishankar%20GB%2C%20Misra%20N%2C%20Holla%20N%2C%20et%20al.%20Chronic%20granulomatous%20disease%2C%20the%20McLeod%20phenotype%20and%20the%20contiguous%20gene%20deletion%20syndrome-a%20review.%20Clin%20Mol%20Allergy%20%5BInternet%5D.%202011%20Nov%2023%20%5Bcited%202022%20Aug%2025%5D%3B9%3A13.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC3267648%5C%2F%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC3267648%5C%2F%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Chronic%20granulomatous%20disease%2C%20the%20McLeod%20phenotype%20and%20the%20contiguous%20gene%20deletion%20syndrome-a%20review%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Casey%20E%22%2C%22lastName%22%3A%22Watkins%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22John%22%2C%22lastName%22%3A%22Litchfield%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eunkyung%22%2C%22lastName%22%3A%22Song%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Gayatri%20B%22%2C%22lastName%22%3A%22Jaishankar%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Niva%22%2C%22lastName%22%3A%22Misra%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nikhil%22%2C%22lastName%22%3A%22Holla%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michelle%22%2C%22lastName%22%3A%22Duffourc%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Guha%22%2C%22lastName%22%3A%22Krishnaswamy%22%7D%5D%2C%22abstractNote%22%3A%22Chronic%20Granulomatous%20Disease%20%28CGD%29%2C%20a%20disorder%20of%20the%20NADPH%20oxidase%20system%2C%20results%20in%20phagocyte%20functional%20defects%20and%20subsequent%20infections%20with%20bacterial%20and%20fungal%20pathogens%20%28such%20as%20Aspergillus%20species%20and%20Candida%20albicans%29.%20Deletions%20and%20missense%2C%20frameshift%2C%20or%20nonsense%20mutations%20in%20the%20gp91phox%20gene%20%28also%20termed%20CYBB%29%2C%20located%20in%20the%20Xp21.1%20region%20of%20the%20X%20chromosome%2C%20are%20associated%20with%20the%20most%20common%20form%20of%20CGD.%20When%20larger%20X-chromosomal%20deletions%20occur%2C%20including%20the%20XK%20gene%20deletion%2C%20a%20so-called%20%26quot%3BContiguous%20Gene%20Deletion%20Syndrome%26quot%3B%20may%20result.%20The%20contiguous%20gene%20deletion%20syndrome%20is%20known%20to%20associate%20the%20Kell%20phenotype%5C%2FMcLeod%20syndrome%20with%20diseases%20such%20as%20X-linked%20chronic%20granulomatous%20disease%2C%20Duchenne%20muscular%20dystrophy%2C%20and%20X-linked%20retinitis%20pigmentosa.%20These%20patients%20are%20often%20complicated%20and%20management%20requires%20special%20attention%20to%20the%20various%20facets%20of%20the%20syndrome.%22%2C%22date%22%3A%222011-11-23%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1186%5C%2F1476-7961-9-13%22%2C%22ISSN%22%3A%221476-7961%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.ncbi.nlm.nih.gov%5C%2Fpmc%5C%2Farticles%5C%2FPMC3267648%5C%2F%22%2C%22collections%22%3A%5B%22CTPGWM38%22%5D%2C%22dateModified%22%3A%222022-08-25T15%3A17%3A52Z%22%7D%7D%2C%7B%22key%22%3A%22PVKFJ6PW%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Weaver%20and%20Christian%22%2C%22parsedDate%22%3A%221980-06%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BWeaver%20DD%2C%20Christian%20JC.%20Familial%20variation%20of%20head%20size%20and%20adjustment%20for%20parental%20head%20circumference.%20J%20Pediatr.%201980%20Jun%3B96%286%29%3A990%26%23x2013%3B4.%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Familial%20variation%20of%20head%20size%20and%20adjustment%20for%20parental%20head%20circumference%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22D.%20D.%22%2C%22lastName%22%3A%22Weaver%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22J.%20C.%22%2C%22lastName%22%3A%22Christian%22%7D%5D%2C%22abstractNote%22%3A%22Occipitofrontal%20head%20circumference%20was%20measured%20on%20122%20twin%20pairs%2C%20their%20spouses%2C%20and%20their%20children.%20These%20data%20indicate%20that%20approximately%2050%25%20of%20normal%20head%20size%20variation%20is%20familial.%20Because%20of%20the%20relationship%20between%20the%20head%20size%20of%20normal%20children%20and%20their%20parents%2C%20adjustment%20of%20a%20child%26%23039%3Bs%20head%20size%20value%20by%20the%20average%20parental%20value%20permits%20better%20definition%20of%20the%20range%20of%20normalcy.%20A%20method%20is%20presented%20that%20will%20allow%20physicians%20to%20make%20this%20adjustment%2C%20providing%20a%20more%20refined%20assessment%20of%20head%20size%20when%20there%20is%20a%20suspected%20abnormality.%22%2C%22date%22%3A%221980-06%22%2C%22language%22%3A%22eng%22%2C%22DOI%22%3A%2210.1016%5C%2Fs0022-3476%2880%2980623-8%22%2C%22ISSN%22%3A%220022-3476%22%2C%22url%22%3A%22%22%2C%22collections%22%3A%5B%22PUNJ3C2T%22%5D%2C%22dateModified%22%3A%222022-08-17T19%3A03%3A04Z%22%7D%7D%2C%7B%22key%22%3A%22MPJDID52%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22P%5Cu00f6s%20et%20al.%22%2C%22parsedDate%22%3A%222021-10-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BP%26%23xF6%3Bs%20O%2C%20Radvanszky%20J%2C%20Bugly%26%23xF3%3B%20G%2C%20P%26%23xF6%3Bs%20Z%2C%20Rusnakova%20D%2C%20Nagy%20B%2C%20et%20al.%20DNA%20copy%20number%20variation%3A%20Main%20characteristics%2C%20evolutionary%20significance%2C%20and%20pathological%20aspects.%20Biomedical%20Journal%20%5BInternet%5D.%202021%20Oct%201%20%5Bcited%202022%20Aug%2017%5D%3B44%285%29%3A548%26%23x2013%3B59.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS2319417021000093%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS2319417021000093%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22DNA%20copy%20number%20variation%3A%20Main%20characteristics%2C%20evolutionary%20significance%2C%20and%20pathological%20aspects%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ondrej%22%2C%22lastName%22%3A%22P%5Cu00f6s%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Jan%22%2C%22lastName%22%3A%22Radvanszky%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Gergely%22%2C%22lastName%22%3A%22Bugly%5Cu00f3%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Zuzana%22%2C%22lastName%22%3A%22P%5Cu00f6s%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Diana%22%2C%22lastName%22%3A%22Rusnakova%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22B%5Cu00e1lint%22%2C%22lastName%22%3A%22Nagy%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Tomas%22%2C%22lastName%22%3A%22Szemes%22%7D%5D%2C%22abstractNote%22%3A%22Copy%20number%20variants%20%28CNVs%29%20were%20the%20subject%20of%20extensive%20research%20in%20the%20past%20years.%20They%20are%20common%20features%20of%20the%20human%20genome%20that%20play%20an%20important%20role%20in%20evolution%2C%20contribute%20to%20population%20diversity%2C%20development%20of%20certain%20diseases%2C%20and%20influence%20host%5Cu2013microbiome%20interactions.%20CNVs%20have%20found%20application%20in%20the%20molecular%20diagnosis%20of%20many%20diseases%20and%20in%20non-invasive%20prenatal%20care%2C%20but%20their%20full%20potential%20is%20only%20emerging.%20CNVs%20are%20expected%20to%20have%20a%20tremendous%20impact%20on%20screening%2C%20diagnosis%2C%20prognosis%2C%20and%20monitoring%20of%20several%20disorders%2C%20including%20cancer%20and%20cardiovascular%20disease.%20Here%2C%20we%20comprehensively%20review%20basic%20definitions%20of%20the%20term%20CNV%2C%20outline%20mechanisms%20and%20factors%20involved%20in%20CNV%20formation%2C%20and%20discuss%20their%20evolutionary%20and%20pathological%20aspects.%20We%20suggest%20a%20need%20for%20better%20defined%20distinguishing%20criteria%20and%20boundaries%20between%20known%20types%20of%20CNVs.%22%2C%22date%22%3A%222021-10-01%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1016%5C%2Fj.bj.2021.02.003%22%2C%22ISSN%22%3A%222319-4170%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.sciencedirect.com%5C%2Fscience%5C%2Farticle%5C%2Fpii%5C%2FS2319417021000093%22%2C%22collections%22%3A%5B%22RJKGI5D3%22%5D%2C%22dateModified%22%3A%222022-08-17T18%3A49%3A49Z%22%7D%7D%2C%7B%22key%22%3A%22WWSKTFP6%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Talukdar%20et%20al.%22%2C%22parsedDate%22%3A%222019-11-01%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BTalukdar%20S%2C%20Hawkes%20L%2C%20Hanson%20H%2C%20Kulkarni%20A%2C%20Brady%20AF%2C%20McMullan%20DJ%2C%20et%20al.%20Structural%20Aberrations%20with%20Secondary%20Implications%20%28SASIs%29%3A%20consensus%20recommendations%20for%20reporting%20of%20cancer%20susceptibility%20genes%20identified%20during%20analysis%20of%20Copy%20Number%20Variants%20%28CNVs%29.%20Journal%20of%20Medical%20Genetics%20%5BInternet%5D.%202019%20Nov%201%20%5Bcited%202022%20Aug%2017%5D%3B56%2811%29%3A718%26%23x2013%3B26.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fjmg.bmj.com%5C%2Fcontent%5C%2F56%5C%2F11%5C%2F718%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fjmg.bmj.com%5C%2Fcontent%5C%2F56%5C%2F11%5C%2F718%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Structural%20Aberrations%20with%20Secondary%20Implications%20%28SASIs%29%3A%20consensus%20recommendations%20for%20reporting%20of%20cancer%20susceptibility%20genes%20identified%20during%20analysis%20of%20Copy%20Number%20Variants%20%28CNVs%29%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sabrina%22%2C%22lastName%22%3A%22Talukdar%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Lara%22%2C%22lastName%22%3A%22Hawkes%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Helen%22%2C%22lastName%22%3A%22Hanson%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Anjana%22%2C%22lastName%22%3A%22Kulkarni%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Angela%20F.%22%2C%22lastName%22%3A%22Brady%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Dominic%20J.%22%2C%22lastName%22%3A%22McMullan%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Joo%20Wook%22%2C%22lastName%22%3A%22Ahn%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Emma%22%2C%22lastName%22%3A%22Woodward%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Clare%22%2C%22lastName%22%3A%22Turnbull%22%7D%5D%2C%22abstractNote%22%3A%22Clinical%20testing%20with%20chromosomal%20microarray%20%28CMA%29%20is%20most%20commonly%20undertaken%20for%20clinical%20indications%20such%20as%20intellectual%20disability%2C%20dysmorphic%20features%20and%5C%2For%20congenital%20abnormalities.%20Identification%20of%20a%20structural%20aberration%20%28SA%29%20involving%20a%20cancer%20susceptibility%20gene%20%28CSG%29%20constitutes%20a%20type%20of%20incidental%20or%20secondary%20finding.%20Laboratory%20reporting%2C%20risk%20communication%20and%20clinical%20management%20of%20these%20structural%20aberrations%20with%20secondary%20implications%20%28SASIs%29%20is%20currently%20inconsistent.%20We%20undertake%20meta-analysis%20of%2018%20622%20instances%20of%20CMA%20performed%20for%20unrelated%20indications%20in%20which%20106%20SASIs%20are%20identified%20involving%20in%20total%2040%20different%20CSGs.%20Here%20we%20present%20the%20recommendations%20of%20a%20joint%20UK%20working%20group%20representing%20the%20British%20Society%20of%20Genomic%20Medicine%2C%20UK%20Cancer%20Genetics%20Group%20and%20UK%20Association%20for%20Clinical%20Genomic%20Science.%20SASIs%20are%20categorised%20into%20four%20groups%2C%20defined%20by%20the%20type%20of%20SA%20and%20the%20cancer%20risk.%20For%20each%20group%2C%20recommendations%20are%20provided%20regarding%20reflex%20parental%20testing%20and%20cancer%20risk%20management.%22%2C%22date%22%3A%222019%5C%2F11%5C%2F01%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1136%5C%2Fjmedgenet-2018-105820%22%2C%22ISSN%22%3A%220022-2593%2C%201468-6244%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fjmg.bmj.com%5C%2Fcontent%5C%2F56%5C%2F11%5C%2F718%22%2C%22collections%22%3A%5B%22TA77IF8F%22%5D%2C%22dateModified%22%3A%222022-08-17T18%3A47%3A24Z%22%7D%7D%2C%7B%22key%22%3A%22XN9ENBJD%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Mossink%20et%20al.%22%2C%22parsedDate%22%3A%222021-03-01%22%2C%22numChildren%22%3A1%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BMossink%20B%2C%20Negwer%20M%2C%20Schubert%20D%2C%20Nadif%20Kasri%20N.%20The%20emerging%20role%20of%20chromatin%20remodelers%20in%20neurodevelopmental%20disorders%3A%20a%20developmental%20perspective.%20Cell%20Mol%20Life%20Sci%20%5BInternet%5D.%202021%20Mar%201%20%5Bcited%202022%20Aug%2015%5D%3B78%286%29%3A2517%26%23x2013%3B63.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2Fs00018-020-03714-5%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2Fs00018-020-03714-5%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22The%20emerging%20role%20of%20chromatin%20remodelers%20in%20neurodevelopmental%20disorders%3A%20a%20developmental%20perspective%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Britt%22%2C%22lastName%22%3A%22Mossink%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Moritz%22%2C%22lastName%22%3A%22Negwer%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Dirk%22%2C%22lastName%22%3A%22Schubert%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nael%22%2C%22lastName%22%3A%22Nadif%20Kasri%22%7D%5D%2C%22abstractNote%22%3A%22Neurodevelopmental%20disorders%20%28NDDs%29%2C%20including%20intellectual%20disability%20%28ID%29%20and%20autism%20spectrum%20disorders%20%28ASD%29%2C%20are%20a%20large%20group%20of%20disorders%20in%20which%20early%20insults%20during%20brain%20development%20result%20in%20a%20wide%20and%20heterogeneous%20spectrum%20of%20clinical%20diagnoses.%20Mutations%20in%20genes%20coding%20for%20chromatin%20remodelers%20are%20overrepresented%20in%20NDD%20cohorts%2C%20pointing%20towards%20epigenetics%20as%20a%20convergent%20pathogenic%20pathway%20between%20these%20disorders.%20In%20this%20review%20we%20detail%20the%20role%20of%20NDD-associated%20chromatin%20remodelers%20during%20the%20developmental%20continuum%20of%20progenitor%20expansion%2C%20differentiation%2C%20cell-type%20specification%2C%20migration%20and%20maturation.%20We%20discuss%20how%20defects%20in%20chromatin%20remodelling%20during%20these%20early%20developmental%20time%20points%20compound%20over%20time%20and%20result%20in%20impaired%20brain%20circuit%20establishment.%20In%20particular%2C%20we%20focus%20on%20their%20role%20in%20the%20three%20largest%20cell%20populations%3A%20glutamatergic%20neurons%2C%20GABAergic%20neurons%2C%20and%20glia%20cells.%20An%20in-depth%20understanding%20of%20the%20spatiotemporal%20role%20of%20chromatin%20remodelers%20during%20neurodevelopment%20can%20contribute%20to%20the%20identification%20of%20molecular%20targets%20for%20treatment%20strategies.%22%2C%22date%22%3A%222021-03-01%22%2C%22language%22%3A%22en%22%2C%22DOI%22%3A%2210.1007%5C%2Fs00018-020-03714-5%22%2C%22ISSN%22%3A%221420-9071%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1007%5C%2Fs00018-020-03714-5%22%2C%22collections%22%3A%5B%227ZJWGN9I%22%5D%2C%22dateModified%22%3A%222022-08-15T10%3A13%3A18Z%22%7D%7D%2C%7B%22key%22%3A%22TL5MNDGV%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Murphy%20et%20al.%22%2C%22parsedDate%22%3A%222006-07-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BMurphy%20NA%2C%20Elias%20ER%2C%20for%20the%20Council%20on%20Children%20With%20Disabilities.%20Sexuality%20of%20Children%20and%20Adolescents%20With%20Developmental%20Disabilities.%20Pediatrics%20%5BInternet%5D.%202006%20Jul%201%20%5Bcited%202022%20Jul%2022%5D%3B118%281%29%3A398%26%23x2013%3B403.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1542%5C%2Fpeds.2006-1115%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1542%5C%2Fpeds.2006-1115%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Sexuality%20of%20Children%20and%20Adolescents%20With%20Developmental%20Disabilities%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nancy%20A.%22%2C%22lastName%22%3A%22Murphy%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ellen%20Roy%22%2C%22lastName%22%3A%22Elias%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22name%22%3A%22for%20the%20Council%20on%20Children%20With%20Disabilities%22%7D%5D%2C%22abstractNote%22%3A%22Children%20and%20adolescents%20with%20developmental%20disabilities%2C%20like%20all%20children%2C%20are%20sexual%20persons.%20However%2C%20attention%20to%20their%20complex%20medical%20and%20functional%20issues%20often%20consumes%20time%20that%20might%20otherwise%20be%20invested%20in%20addressing%20the%20anatomic%2C%20physiologic%2C%20emotional%2C%20and%20social%20aspects%20of%20their%20developing%20sexuality.%20This%20report%20discusses%20issues%20of%20puberty%2C%20contraception%2C%20psychosexual%20development%2C%20sexual%20abuse%2C%20and%20sexuality%20education%20specific%20to%20children%20and%20adolescents%20with%20disabilities%20and%20their%20families.%20Pediatricians%2C%20in%20the%20context%20of%20the%20medical%20home%2C%20are%20encouraged%20to%20discuss%20issues%20of%20sexuality%20on%20a%20regular%20basis%2C%20ensure%20the%20privacy%20of%20each%20child%20and%20adolescent%2C%20promote%20self-care%20and%20social%20independence%20among%20persons%20with%20disabilities%2C%20advocate%20for%20appropriate%20sexuality%20education%2C%20and%20provide%20ongoing%20education%20for%20children%20and%20adolescents%20with%20developmental%20disabilities%20and%20their%20families.%22%2C%22date%22%3A%222006-07-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1542%5C%2Fpeds.2006-1115%22%2C%22ISSN%22%3A%220031-4005%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1542%5C%2Fpeds.2006-1115%22%2C%22collections%22%3A%5B%22PUNJ3C2T%22%5D%2C%22dateModified%22%3A%222022-07-22T07%3A55%3A26Z%22%7D%7D%2C%7B%22key%22%3A%22MDPZVGHQ%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Quint%20et%20al.%22%2C%22parsedDate%22%3A%222016-07-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BQuint%20EH%2C%20O%26%23x2019%3BBrien%20RF%2C%20COMMITTEE%20ON%20ADOLESCENCE%2C%20The%20North%20American%20Society%20for%20Pediatric%20and%20Adolescent%20Gynecology%2C%20Braverman%20PK%2C%20Adelman%20WP%2C%20et%20al.%20Menstrual%20Management%20for%20Adolescents%20With%20Disabilities.%20Pediatrics%20%5BInternet%5D.%202016%20Jul%201%20%5Bcited%202022%20Jul%2022%5D%3B138%281%29%3Ae20160295.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1542%5C%2Fpeds.2016-0295%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1542%5C%2Fpeds.2016-0295%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Menstrual%20Management%20for%20Adolescents%20With%20Disabilities%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Elisabeth%20H.%22%2C%22lastName%22%3A%22Quint%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rebecca%20F.%22%2C%22lastName%22%3A%22O%5Cu2019Brien%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22name%22%3A%22COMMITTEE%20ON%20ADOLESCENCE%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22name%22%3A%22The%20North%20American%20Society%20for%20Pediatric%20and%20Adolescent%20Gynecology%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Paula%20K.%22%2C%22lastName%22%3A%22Braverman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22William%20P.%22%2C%22lastName%22%3A%22Adelman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Elizabeth%20M.%22%2C%22lastName%22%3A%22Alderman%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Cora%20C.%22%2C%22lastName%22%3A%22Breuner%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22David%20A.%22%2C%22lastName%22%3A%22Levine%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Arik%20V.%22%2C%22lastName%22%3A%22Marcell%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Rebecca%20F.%22%2C%22lastName%22%3A%22O%5Cu2019Brien%22%7D%5D%2C%22abstractNote%22%3A%22The%20onset%20of%20menses%20for%20adolescents%20with%20physical%20or%20intellectual%20disabilities%20can%20affect%20their%20independence%20and%20add%20additional%20concerns%20for%20families%20at%20home%2C%20in%20schools%2C%20and%20in%20other%20settings.%20The%20pediatrician%20is%20the%20primary%20health%20care%20provider%20to%20explore%20and%20assist%20with%20the%20pubertal%20transition%20and%20menstrual%20management.%20Menstrual%20management%20of%20both%20normal%20and%20abnormal%20cycles%20may%20be%20requested%20to%20minimize%20hygiene%20issues%2C%20premenstrual%20symptoms%2C%20dysmenorrhea%2C%20heavy%20or%20irregular%20bleeding%2C%20contraception%2C%20and%20conditions%20exacerbated%20by%20the%20menstrual%20cycle.%20Several%20options%20are%20available%20for%20menstrual%20management%2C%20depending%20on%20the%20outcome%20that%20is%20desired%2C%20ranging%20from%20cycle%20regulation%20to%20complete%20amenorrhea.%20The%20use%20of%20medications%20or%20the%20request%20for%20surgeries%20to%20help%20with%20the%20menstrual%20cycles%20in%20teenagers%20with%20disabilities%20has%20medical%2C%20social%2C%20legal%2C%20and%20ethical%20implications.%20This%20clinical%20report%20is%20designed%20to%20help%20guide%20pediatricians%20in%20assisting%20adolescent%20females%20with%20intellectual%20and%5C%2For%20physical%20disabilities%20and%20their%20families%20in%20making%20decisions%20related%20to%20successfully%20navigating%20menarche%20and%20subsequent%20menstrual%20cycles.%22%2C%22date%22%3A%222016-07-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1542%5C%2Fpeds.2016-0295%22%2C%22ISSN%22%3A%220031-4005%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fdoi.org%5C%2F10.1542%5C%2Fpeds.2016-0295%22%2C%22collections%22%3A%5B%22PUNJ3C2T%22%5D%2C%22dateModified%22%3A%222022-07-22T07%3A55%3A23Z%22%7D%7D%2C%7B%22key%22%3A%228WB4WP4U%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22McHugh%20et%20al.%22%2C%22parsedDate%22%3A%222021-03-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BMcHugh%20JC%2C%20Daly%20N%2C%20Colfer%20A.%20Measuring%20the%20effects%20of%20pre-test%20probability%20on%20out-patient%20first%20EEG%20investigation%20in%20children%20%26%23x2013%3B%20A%20guide%20to%20evidence-based%20EEG%20triage%20in%20a%20pandemic.%20Seizure%20-%20European%20Journal%20of%20Epilepsy%20%5BInternet%5D.%202021%20Mar%201%20%5Bcited%202022%20Jul%2020%5D%3B86%3A8%26%23x2013%3B15.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttps%3A%5C%2F%5C%2Fwww.seizure-journal.com%5C%2Farticle%5C%2FS1059-1311%252821%252900010-8%5C%2Ffulltext%26%23039%3B%26gt%3Bhttps%3A%5C%2F%5C%2Fwww.seizure-journal.com%5C%2Farticle%5C%2FS1059-1311%252821%252900010-8%5C%2Ffulltext%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22Measuring%20the%20effects%20of%20pre-test%20probability%20on%20out-patient%20first%20EEG%20investigation%20in%20children%20%5Cu2013%20A%20guide%20to%20evidence-based%20EEG%20triage%20in%20a%20pandemic%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22John%20C.%22%2C%22lastName%22%3A%22McHugh%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Nicole%22%2C%22lastName%22%3A%22Daly%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Ailish%22%2C%22lastName%22%3A%22Colfer%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222021-03-01%22%2C%22language%22%3A%22English%22%2C%22DOI%22%3A%2210.1016%5C%2Fj.seizure.2021.01.007%22%2C%22ISSN%22%3A%221059-1311%2C%201532-2688%22%2C%22url%22%3A%22https%3A%5C%2F%5C%2Fwww.seizure-journal.com%5C%2Farticle%5C%2FS1059-1311%252821%252900010-8%5C%2Ffulltext%22%2C%22collections%22%3A%5B%225Z9YTE43%22%5D%2C%22dateModified%22%3A%222022-07-20T09%3A27%3A43Z%22%7D%7D%2C%7B%22key%22%3A%228AZB46WW%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Koutroumanidis%20et%20al.%22%2C%22parsedDate%22%3A%222017-09-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKoutroumanidis%20M%2C%20Arzimanoglou%20A%2C%20Caraballo%20R%2C%20Goyal%20S%2C%20Kaminska%20A%2C%20Laoprasert%20P%2C%20et%20al.%20The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%201%29.%20Epileptic%20Disorders%20%5BInternet%5D.%202017%20Sep%201%20%5Bcited%202022%20Jul%2020%5D%3B19%283%29%3A233%26%23x2013%3B98.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508%5C%2Farticle.phtml%3Ftab%3Dtexte%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508%5C%2Farticle.phtml%3Ftab%3Dtexte%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%201%29%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michalis%22%2C%22lastName%22%3A%22Koutroumanidis%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alexis%22%2C%22lastName%22%3A%22Arzimanoglou%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Roberto%22%2C%22lastName%22%3A%22Caraballo%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sushma%22%2C%22lastName%22%3A%22Goyal%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Anna%22%2C%22lastName%22%3A%22Kaminska%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pramote%22%2C%22lastName%22%3A%22Laoprasert%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Hirokazu%22%2C%22lastName%22%3A%22Oguni%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Guido%22%2C%22lastName%22%3A%22Rubboli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22William%22%2C%22lastName%22%3A%22Tatum%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pierre%22%2C%22lastName%22%3A%22Thomas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eugen%22%2C%22lastName%22%3A%22Trinka%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Luca%22%2C%22lastName%22%3A%22Vignatelli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Solomon%20L.%22%2C%22lastName%22%3A%22Mosh%5Cu00e9%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222017-09-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1684%5C%2Fepd.2017.0935%22%2C%22ISSN%22%3A%221294-9361%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508%5C%2Farticle.phtml%3Ftab%3Dtexte%22%2C%22collections%22%3A%5B%225Z9YTE43%22%5D%2C%22dateModified%22%3A%222022-07-20T09%3A11%3A54Z%22%7D%7D%2C%7B%22key%22%3A%22HRZJ846F%22%2C%22library%22%3A%7B%22id%22%3A332710%7D%2C%22meta%22%3A%7B%22creatorSummary%22%3A%22Koutroumanidis%20et%20al.%22%2C%22parsedDate%22%3A%222017-12-01%22%2C%22numChildren%22%3A2%7D%2C%22bib%22%3A%22%26lt%3Bdiv%20class%3D%26quot%3Bcsl-bib-body%26quot%3B%20style%3D%26quot%3Bline-height%3A%201.35%3B%20%26quot%3B%26gt%3B%5Cn%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-entry%26quot%3B%20style%3D%26quot%3Bclear%3A%20left%3B%20%26quot%3B%26gt%3B%5Cn%20%20%20%20%26lt%3Bdiv%20class%3D%26quot%3Bcsl-left-margin%26quot%3B%20style%3D%26quot%3Bfloat%3A%20left%3B%20padding-right%3A%200.5em%3B%20text-align%3A%20right%3B%20width%3A%201em%3B%26quot%3B%26gt%3B1.%20%26lt%3B%5C%2Fdiv%26gt%3B%26lt%3Bdiv%20class%3D%26quot%3Bcsl-right-inline%26quot%3B%20style%3D%26quot%3Bmargin%3A%200%20.4em%200%201.5em%3B%26quot%3B%26gt%3BKoutroumanidis%20M%2C%20Arzimanoglou%20A%2C%20Caraballo%20R%2C%20Goyal%20S%2C%20Kaminska%20A%2C%20Laoprasert%20P%2C%20et%20al.%20The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%202%29.%20Epileptic%20Disorders%20%5BInternet%5D.%202017%20Dec%201%20%5Bcited%202022%20Jul%2020%5D%3B19%284%29%3A385%26%23x2013%3B437.%20Available%20from%3A%20%26lt%3Ba%20class%3D%26%23039%3Bzp-ItemURL%26%23039%3B%20href%3D%26%23039%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218%5C%2Farticle.phtml%3Ftab%3Dtexte%26%23039%3B%26gt%3Bhttp%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218%5C%2Farticle.phtml%3Ftab%3Dtexte%26lt%3B%5C%2Fa%26gt%3B%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%20%20%26lt%3B%5C%2Fdiv%26gt%3B%5Cn%26lt%3B%5C%2Fdiv%26gt%3B%22%2C%22data%22%3A%7B%22itemType%22%3A%22journalArticle%22%2C%22title%22%3A%22The%20role%20of%20EEG%20in%20the%20diagnosis%20and%20classification%20of%20the%20epilepsy%20syndromes%3A%20a%20tool%20for%20clinical%20practice%20by%20the%20ILAE%20Neurophysiology%20Task%20Force%20%28Part%202%29%22%2C%22creators%22%3A%5B%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Michalis%22%2C%22lastName%22%3A%22Koutroumanidis%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Alexis%22%2C%22lastName%22%3A%22Arzimanoglou%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Roberto%22%2C%22lastName%22%3A%22Caraballo%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Sushma%22%2C%22lastName%22%3A%22Goyal%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Anna%22%2C%22lastName%22%3A%22Kaminska%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pramote%22%2C%22lastName%22%3A%22Laoprasert%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Hirokazu%22%2C%22lastName%22%3A%22Oguni%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Guido%22%2C%22lastName%22%3A%22Rubboli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22William%22%2C%22lastName%22%3A%22Tatum%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Pierre%22%2C%22lastName%22%3A%22Thomas%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Eugen%22%2C%22lastName%22%3A%22Trinka%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Luca%22%2C%22lastName%22%3A%22Vignatelli%22%7D%2C%7B%22creatorType%22%3A%22author%22%2C%22firstName%22%3A%22Solomon%20L.%22%2C%22lastName%22%3A%22Mosh%5Cu00e9%22%7D%5D%2C%22abstractNote%22%3A%22%22%2C%22date%22%3A%222017-12-01%22%2C%22language%22%3A%22%22%2C%22DOI%22%3A%2210.1684%5C%2Fepd.2017.0952%22%2C%22ISSN%22%3A%221294-9361%22%2C%22url%22%3A%22http%3A%5C%2F%5C%2Fwww.jle.com%5C%2Ffr%5C%2Frevues%5C%2Fepd%5C%2Fe-docs%5C%2Fthe_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218%5C%2Farticle.phtml%3Ftab%3Dtexte%22%2C%22collections%22%3A%5B%225Z9YTE43%22%5D%2C%22dateModified%22%3A%222022-07-20T09%3A11%3A48Z%22%7D%7D%5D%7D
1.
Specchio N, Wirrell EC, Scheffer IE, Nabbout R, Riney K, Samia P, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia [Internet]. 2022 [cited 2022 Oct 22];63(6):1398–442. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/epi.17241
1.
Kang B, Kwon YS. Benign convulsion with mild gastroenteritis. Korean J Pediatr [Internet]. 2014 [cited 2022 Oct 22];57(7):304. Available from: http://www.e-cep.org/journal/view.php?doi=10.3345/kjp.2014.57.7.304
1.
Lee YS, Lee GH, Kwon YS. Update on benign convulsions with mild gastroenteritis. Clin Exp Pediatr [Internet]. 2021 Dec 27 [cited 2022 Oct 22];65(10):469–75. Available from: http://www.e-cep.org/journal/view.php?doi=10.3345/cep.2021.00997
1.
Verity C, Baker E, Maunder P, Pal S, Winstone AM. Differential diagnosis of progressive intellectual and neurological deterioration in children. Dev Med Child Neurol [Internet]. 2021 Mar [cited 2022 Oct 15];63(3):287–94. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7891454/
1.
Hoffmann K, Müller JS, Stricker S, Megarbane A, Rajab A, Lindner TH, et al. Escobar Syndrome Is a Prenatal Myasthenia Caused by Disruption of the Acetylcholine Receptor Fetal γ Subunit. The American Journal of Human Genetics [Internet]. 2006 Aug 1 [cited 2022 Oct 15];79(2):303–12. Available from: https://www.sciencedirect.com/science/article/pii/S0002929707631371
1.
Hacohen Y, Jacobson LW, Byrne S, Norwood F, Lall A, Robb S, et al. Fetal acetylcholine receptor inactivation syndrome: A myopathy due to maternal antibodies. Neurology - Neuroimmunology Neuroinflammation [Internet]. 2015 Feb 1 [cited 2022 Oct 15];2(1). Available from: https://nn.neurology.org/content/2/1/e57
1.
Polyak A, Kubina RM, Girirajan S. Comorbidity of intellectual disability confounds ascertainment of autism: implications for genetic diagnosis. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics [Internet]. 2015 [cited 2022 Oct 15];168(7):600–8. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.b.32338
1.
Moore CA, Staples JE, Dobyns WB, Pessoa A, Ventura CV, Fonseca EB da, et al. Characterizing the Pattern of Anomalies in Congenital Zika Syndrome for Pediatric Clinicians. JAMA Pediatrics [Internet]. 2017 Mar 1 [cited 2022 Oct 15];171(3):288–95. Available from: https://doi.org/10.1001/jamapediatrics.2016.3982
1.
Adebanjo T. Update: Interim Guidance for the Diagnosis, Evaluation, and Management of Infants with Possible Congenital Zika Virus Infection — United States, October 2017. MMWR Morb Mortal Wkly Rep [Internet]. 2017 [cited 2022 Oct 15];66. Available from: https://www.cdc.gov/mmwr/volumes/66/wr/mm6641a1.htm
1.
Koens LH, Tijssen MAJ, Lange F, Wolffenbuttel BHR, Rufa A, Zee DS, et al. Eye movement disorders and neurological symptoms in late‐onset inborn errors of metabolism. Mov Disord [Internet]. 2018 Dec [cited 2022 Oct 10];33(12):1844–56. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6587951/
1.
Brodsky MC. Pediatric Neuro-Ophthalmology [Internet]. New York, NY: Springer; 2010 [cited 2022 Oct 10]. Available from: http://link.springer.com/10.1007/978-0-387-69069-8
1.
Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, et al. Recurrence Risk for Autism Spectrum Disorders: A Baby Siblings Research Consortium Study. Pediatrics [Internet]. 2011 Sep [cited 2022 Oct 10];128(3):e488–95. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3164092/
1.
Yoo J. Differential diagnosis and management of hyperpigmentation. Clinical and Experimental Dermatology [Internet]. 2022 [cited 2022 Oct 8];47(2):251–8. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/ced.14747
1.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics [Internet]. 2019 Jun 1 [cited 2022 Oct 3];35(11):1978–80. Available from: https://doi.org/10.1093/bioinformatics/bty897
1.
Tornese G, Pellegrin MC, Barbi E, Ventura A. Pediatric endocrinology through syndromes. European Journal of Medical Genetics [Internet]. 2020 [cited 2022 Oct 3];63(1):103614. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1769721218303161
1.
Annual Health Checks for People with Intellectual Disabilities in General Practice [Internet]. [cited 2022 Oct 3]. Available from: http://www.intellectualdisability.info/how-to-guides/articles/annual-health-checks-for-people-with-intellectual-disabilities-in-general-practice
1.
Lagorio I, Brunelli L, Striano P. Paroxysmal Nonepileptic Events in Children: A Video Gallery and a Guide for Differential Diagnosis. Neurology: Clinical Practice [Internet]. 2022 Aug 1 [cited 2022 Oct 3];12(4):320–7. Available from: https://cp.neurology.org/content/12/4/320
1.
de Freitas FD, Pimenta S, Soares S, Gonzaga D, Vaz-Matos I, Prior C. El papel de los cannabinoides en los trastornos del neurodesarrollo de niños y adolescentes. Rev Neurol. :9.
1.
Marwaha S, Knowles JW, Ashley EA. A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Medicine [Internet]. 2022 Feb 28 [cited 2022 Oct 3];14(1):23. Available from: https://doi.org/10.1186/s13073-022-01026-w
1.
Patel RA, Hall DA, Eichenseer S, Bailey M. Movement Disorders and Hematologic Diseases. Mov Disord Clin Pract [Internet]. 2020 Dec 29 [cited 2022 Oct 3];8(2):193–207. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7853188/
1.
Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypic Heterogeneity of Genomic Disorders and Rare Copy-Number Variants. New England Journal of Medicine [Internet]. 2012 Oct 4 [cited 2022 Oct 3];367(14):1321–31. Available from: https://doi.org/10.1056/NEJMoa1200395
1.
Bharath R, Unnikrishnan AG, Thampy MV, Anilkumar A, Nisha B, Praveen VP, et al. Turner syndrome and its variants. Indian J Pediatr [Internet]. 2010 [cited 2022 Sep 24];77(2):193–5. Available from: http://link.springer.com/10.1007/s12098-009-0226-7
1.
Liehr T. Cytogenetically visible copy number variations (CG-CNVs) in banding and molecular cytogenetics of human; about heteromorphisms and euchromatic variants. Mol Cytogenet [Internet]. 2016 Jan 22 [cited 2022 Sep 24];9:5. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4724132/
1.
Nothing’s for sure, that’s for sure: Evaluating variants of uncertain significance | Beyond the Ion Channel [Internet]. [cited 2022 Sep 22]. Available from: http://epilepsygenetics.net/2016/08/11/nothings-for-sure-thats-for-sure-evaluating-variants-of-uncertain-significance/
1.
Vaz-Drago R, Custódio N, Carmo-Fonseca M. Deep intronic mutations and human disease. Hum Genet [Internet]. 2017 [cited 2020 May 3];136(9):1093–111. Available from: http://link.springer.com/10.1007/s00439-017-1809-4
1.
Barros FS, Marussi VHR, Amaral LLF, da Rocha AJ, Campos CMS, Freitas LF, et al. The Rare Neurocutaneous Disorders: Update on Clinical, Molecular, and Neuroimaging Features. Topics in Magnetic Resonance Imaging [Internet]. 2018 [cited 2019 Apr 28];27(6):433–62. Available from: http://Insights.ovid.com/crossref?an=00002142-201812000-00004
1.
Gabriele M, Lopez Tobon A, D’Agostino G, Testa G. The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Progress in Neuro-Psychopharmacology and Biological Psychiatry [Internet]. 2018 Jun 8 [cited 2019 Oct 5];84:306–27. Available from: http://www.sciencedirect.com/science/article/pii/S0278584617305389
1.
Guerra J, Cacabelos R. Genomics of speech and language disorders. JTGG [Internet]. 2019 [cited 2020 Sep 25]; Available from: https://jtggjournal.com/article/view/3118
1.
Truty R, Paul J, Kennemer M, Lincoln SE, Olivares E, Nussbaum RL, et al. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med [Internet]. 2019 [cited 2019 Sep 3];21(1):114–23. Available from: http://www.nature.com/articles/s41436-018-0033-5
1.
Garone G, Capuano A, Travaglini L, Graziola F, Stregapede F, Zanni G, et al. Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias. IJMS [Internet]. 2020 May 20 [cited 2021 Sep 2];21(10):3603. Available from: https://www.mdpi.com/1422-0067/21/10/3603
1.
Karbassi I, Maston GA, Love A, DiVincenzo C, Braastad CD, Elzinga CD, et al. A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders. Human Mutation [Internet]. 2016 [cited 2022 Sep 22];37(1):127–34. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/humu.22918
1.
Beetz C, Bauer P. Dual genetic diagnoses - underappreciated “double trouble.” JBCGenetics [Internet]. 2020 [cited 2022 Sep 13];52–3. Available from: https://www.ejmanager.com/fulltextpdf.php?mno=135141
1.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. New England Journal of Medicine [Internet]. 2017 Jan 5 [cited 2022 Sep 13];376(1):21–31. Available from: https://doi.org/10.1056/NEJMoa1516767
1.
Koutroumanidis M, Arzimanoglou A, Caraballo R, Goyal S, Kaminska A, Laoprasert P, et al. The role of EEG in the diagnosis and classification of the epilepsy syndromes: a tool for clinical practice by the ILAE Neurophysiology Task Force (Part 2). Epileptic Disorders [Internet]. 2017 Dec 1 [cited 2022 Sep 12];19(4):385–437. Available from: http://www.jle.com/fr/revues/epd/e-docs/the_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218/article.phtml?tab=texte
1.
Koutroumanidis M, Arzimanoglou A, Caraballo R, Goyal S, Kaminska A, Laoprasert P, et al. The role of EEG in the diagnosis and classification of the epilepsy syndromes: a tool for clinical practice by the ILAE Neurophysiology Task Force (Part 1). Epileptic Disorders [Internet]. 2017 Sep 1 [cited 2022 Sep 12];19(3):233–98. Available from: http://www.jle.com/fr/revues/epd/e-docs/the_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508/article.phtml?tab=texte
1.
Bronkhorst AJ, Ungerer V, Oberhofer A, Gabriel S, Polatoglou E, Randeu H, et al. New Perspectives on the Importance of Cell-Free DNA Biology. Diagnostics [Internet]. 2022 Sep 3 [cited 2022 Sep 12];12(9):2147. Available from: https://www.mdpi.com/2075-4418/12/9/2147
1.
Friedman JM. Neurofibromatosis 1 [Internet]. University of Washington, Seattle; 2022 [cited 2022 Sep 12]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1109/
1.
Myths of Human Genetics: Introduction [Internet]. [cited 2022 Sep 6]. Available from: https://udel.edu/~mcdonald/mythintro.html
1.
Zafar S, Doria J, Karceski S. Should we standardize the EEG classification of mild, moderate, and severe cerebral dysfunction? Epilepsy Behav [Internet]. 2020 Nov 1 [cited 2022 Sep 6];112. Available from: https://www.epilepsybehavior.com/article/S1525-5050%2820%2930511-4/fulltext
1.
Marks K, Coutinho E, Vincent A. Maternal-Autoantibody-Related (MAR) Autism: Identifying Neuronal Antigens and Approaching Prospects for Intervention. JCM [Internet]. 2020 Aug 7 [cited 2022 Aug 31];9(8):2564. Available from: https://www.mdpi.com/2077-0383/9/8/2564
1.
Watkins CE, Litchfield J, Song E, Jaishankar GB, Misra N, Holla N, et al. Chronic granulomatous disease, the McLeod phenotype and the contiguous gene deletion syndrome-a review. Clin Mol Allergy [Internet]. 2011 Nov 23 [cited 2022 Aug 25];9:13. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3267648/
1.
Weaver DD, Christian JC. Familial variation of head size and adjustment for parental head circumference. J Pediatr. 1980 Jun;96(6):990–4.
1.
Pös O, Radvanszky J, Buglyó G, Pös Z, Rusnakova D, Nagy B, et al. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomedical Journal [Internet]. 2021 Oct 1 [cited 2022 Aug 17];44(5):548–59. Available from: https://www.sciencedirect.com/science/article/pii/S2319417021000093
1.
Talukdar S, Hawkes L, Hanson H, Kulkarni A, Brady AF, McMullan DJ, et al. Structural Aberrations with Secondary Implications (SASIs): consensus recommendations for reporting of cancer susceptibility genes identified during analysis of Copy Number Variants (CNVs). Journal of Medical Genetics [Internet]. 2019 Nov 1 [cited 2022 Aug 17];56(11):718–26. Available from: https://jmg.bmj.com/content/56/11/718
1.
Mossink B, Negwer M, Schubert D, Nadif Kasri N. The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell Mol Life Sci [Internet]. 2021 Mar 1 [cited 2022 Aug 15];78(6):2517–63. Available from: https://doi.org/10.1007/s00018-020-03714-5
1.
Murphy NA, Elias ER, for the Council on Children With Disabilities. Sexuality of Children and Adolescents With Developmental Disabilities. Pediatrics [Internet]. 2006 Jul 1 [cited 2022 Jul 22];118(1):398–403. Available from: https://doi.org/10.1542/peds.2006-1115
1.
Quint EH, O’Brien RF, COMMITTEE ON ADOLESCENCE, The North American Society for Pediatric and Adolescent Gynecology, Braverman PK, Adelman WP, et al. Menstrual Management for Adolescents With Disabilities. Pediatrics [Internet]. 2016 Jul 1 [cited 2022 Jul 22];138(1):e20160295. Available from: https://doi.org/10.1542/peds.2016-0295
1.
McHugh JC, Daly N, Colfer A. Measuring the effects of pre-test probability on out-patient first EEG investigation in children – A guide to evidence-based EEG triage in a pandemic. Seizure - European Journal of Epilepsy [Internet]. 2021 Mar 1 [cited 2022 Jul 20];86:8–15. Available from: https://www.seizure-journal.com/article/S1059-1311%2821%2900010-8/fulltext
1.
Koutroumanidis M, Arzimanoglou A, Caraballo R, Goyal S, Kaminska A, Laoprasert P, et al. The role of EEG in the diagnosis and classification of the epilepsy syndromes: a tool for clinical practice by the ILAE Neurophysiology Task Force (Part 1). Epileptic Disorders [Internet]. 2017 Sep 1 [cited 2022 Jul 20];19(3):233–98. Available from: http://www.jle.com/fr/revues/epd/e-docs/the_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_1__310508/article.phtml?tab=texte
1.
Koutroumanidis M, Arzimanoglou A, Caraballo R, Goyal S, Kaminska A, Laoprasert P, et al. The role of EEG in the diagnosis and classification of the epilepsy syndromes: a tool for clinical practice by the ILAE Neurophysiology Task Force (Part 2). Epileptic Disorders [Internet]. 2017 Dec 1 [cited 2022 Jul 20];19(4):385–437. Available from: http://www.jle.com/fr/revues/epd/e-docs/the_role_of_eeg_in_the_diagnosis_and_classification_of_the_epilepsy_syndromes_a_tool_for_clinical_practice_by_the_ilae_neurophysiology_task_force_part_2__311218/article.phtml?tab=texte